scRNA-seq 基本分析流程
这篇博客主要个人使用scanpy进行10X scRNA-seq数据的基本分析流程。分析使用的数据是10X multiome产生的的小鼠海马scRNA-seq数据,数据目前未公开。如果想要尝试博客中的一些分析,可以使用公开的小鼠海马单细胞数据。另外multiome产生的数据,通常质量会比单模态的scRNA-seq数据要差一些,所以数据中counts不是特别高也是合理的。
数据准备
在cellranger完成对.fastq测序数据的处理后,会生成如下的三个文件
后续的分析会基于这三个文件。其中barcodes.tsv.gz记录了样本名称(cell, 严格来讲barcodes记录的是包裹细胞的油滴名称,但理想情况下油滴中只有一个细胞), feature.tsv.gz中记录了特征名称(gene), matrix.mtx.gz是UMI(unique molecular identifier) count matrix.
首先使用scanpy读取数据
import numpy as np
import pandas as pd
import scanpy as sc
import os
import sys
sys.path.append('code')
sys.path.append("marker file")
import matplotlib.pyplot as plt
import utils # 自己分析时写的一些函数
sc.settings.verbosity = 2 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor='white')
mus = sc.read_10x_mtx(
'data/MHM/', # the directory with the `.mtx` file
var_names='gene_symbols', # use gene symbols for the variable names (variables-axis index)
cache=True)
mus.var_names_make_unique()
raw_mus = mus.copy() ### 后面cluster注释会用到
读取的数据会以一种特殊的数据格式anndata存储,可以简单理解为一种方便单细胞数据分析的字典。
mus
输出
AnnData object with n_obs × n_vars = 8572 × 32285
var: 'gene_ids', 'feature_types'
可以看到数据中包含8572个细胞,32285个基因。
QC(quality control)
在QC步骤中,主要基于一些简单的统计指标,删除掉一些质量较差的细胞或者基因,减少数据中的噪音。
utils.stage_1_QC(mus)
这里我把一些QC相关函数都写到stage_1_QC函数中了,让整个分析的notebook看起来更简洁,stage_1_QC函数如下
### perform stage_1_QC and visualization
def stage_1_QC(adata):
print(' original data shape ' + str(adata.shape))
print(' Filter out low quality cells and genes' + '===='*10)
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
print(' processed data shape' + str(adata.shape))
print('QC metric calculation' + '===='*10)
adata.var['mt'] = adata.var_names.str.startswith('mt-') # annotate the group of mitochondrial genes as 'mt'
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
print('Stage 1 visualization' + '===='*10)
fig = plt.figure(figsize=(15, 10))
grid = plt.GridSpec(2, 3, hspace=0.4, wspace=0.4)
ax_1 = fig.add_subplot(grid[0,0])
sc.pl.highest_expr_genes(adata, ax = ax_1, n_top=20,show = False)
ax_1.set_title('Highest_expr_genes')
ax_2 = fig.add_subplot(grid[0,1])
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt',ax = ax_2,show=False) # check 线粒体基因情况 无线粒体基因
ax_3 = fig.add_subplot(grid[0,2])
ax_3 = sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts',ax = ax_3, show=False)
ax_4 = fig.add_subplot(grid[1,0])
sc.pl.violin(adata, keys = 'n_genes_by_counts',
jitter=0.4, multi_panel=False,ax=ax_4,show=False)
ax_5 = fig.add_subplot(grid[1,1])
sc.pl.violin(adata, keys = 'total_counts',
jitter=0.4, multi_panel=False,ax=ax_5,show=False)
ax_6 = fig.add_subplot(grid[1,2])
sc.pl.violin(adata, keys = 'pct_counts_mt',
jitter=0.4, multi_panel=False,ax=ax_6,show=False)
plt.show()
可以看到其中核心的操作是
sc.pp.filter_cells(adata, min_genes=200) # 过滤掉表达基因数目低于200的细胞
sc.pp.filter_genes(adata, min_cells=3) # 过滤掉在少于3个细胞中表达的基因
adata.var['mt'] = adata.var_names.str.startswith('mt-') # 将线粒体基因注释出来,线粒体基因通常开头都是mt或者MT,线粒体Mitochondria
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True) # 计算一些QC指标
- 过滤掉表达基因数目低于min_genes的细胞
- 过滤掉在少于三个细胞中表达的基因
- 标记出基因中的线粒体基因,后续会删掉线粒体基因比例过高的细胞。
- 计算一些常见的QC指标。
这里过滤细胞和基因的阈值要根据分析具体设定,比如你的细胞数目很少的时候,继续使用’min_genes=200’就不合适;或者当数据中包含中性粒细胞时,由于中性粒细胞是低UMIcount的一类细胞,所以要恰当的降低min_genes的阈值。
对线粒体基因表达比例较高的细胞,通常认为是死亡的细胞,一般会删除。但同样也有例外,心肌细胞、肿瘤细胞就是天然线粒体基因比例高的细胞。因此也要根据后续分析的结果去调整线粒体删选的阈值。
完成第一阶段QC后,我们通常会可视化一些指标,从而人为的设定后续的一些QC阈值。
输出
original data shape (8572, 32285)
Filter out low quality cells and genes========================================
filtered out 117 cells that have less than 200 genes expressed
filtered out 10113 genes that are detected in less than 3 cells
processed data shape(8455, 22172)
QC metric calculation========================================
Stage 1 visualization========================================
normalizing counts per cell
finished (0:00:00)
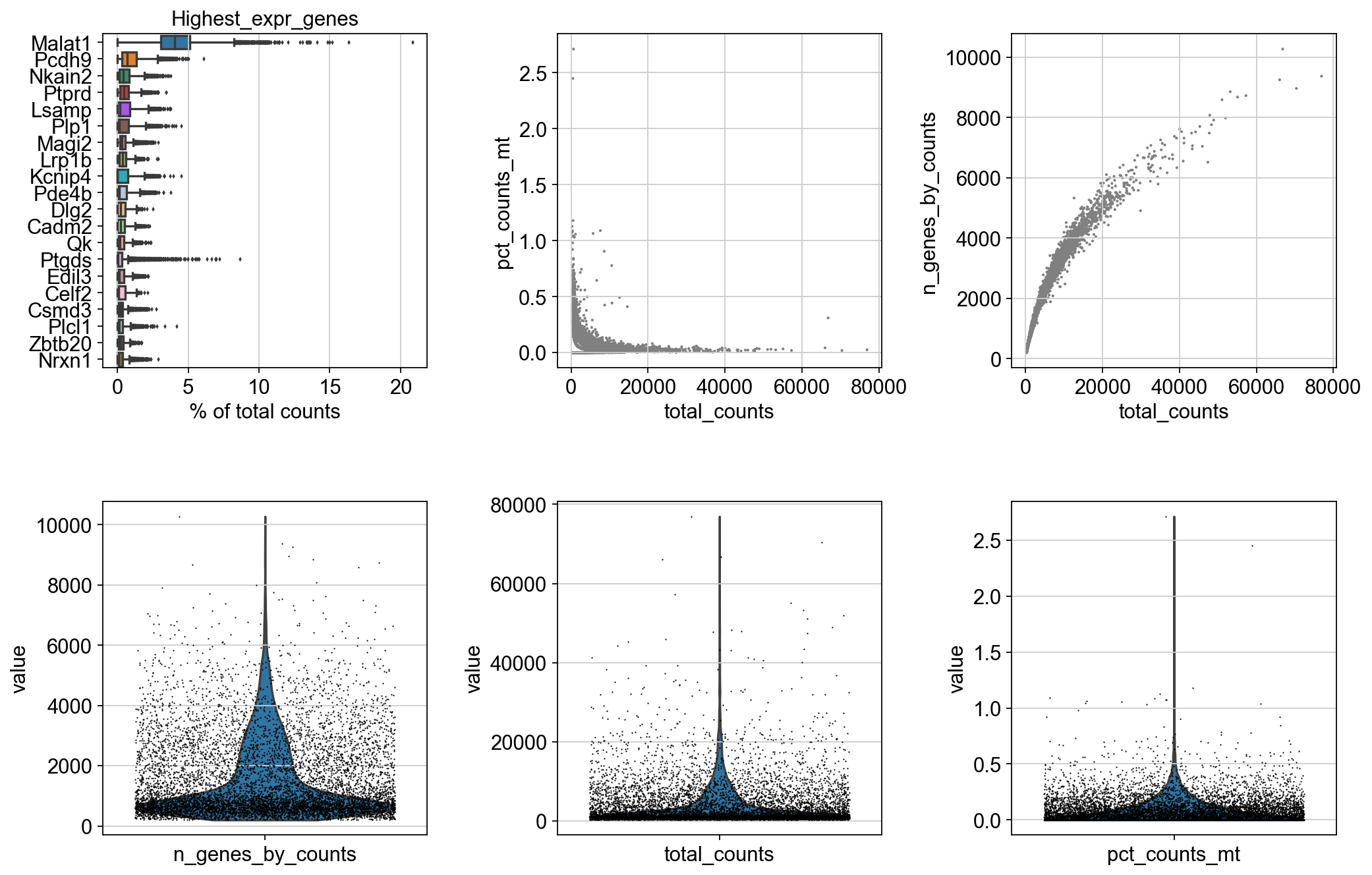
图2中Highest_expr_genes记录的是在所有细胞中表达最高的几个基因,这些基因通常在各个组织中都是高表达的,例如Malat1。因此如果在该图中找不到常见的高表达基因,就需要再次确认数据质量了。
pct_counts_mt 记录了各个细胞中线粒体基因表达比例,n_genes_by_counts 记录了各个细胞中有表达的基因数目。下面的三个小提琴图也是记录的这些信息。结合这些图我们看到,pct_counts_mt > 1.0, n_genes_by_counts >8000的细胞更可能是离群点,删掉。
# 根据 total_counts - n_genes_by_counts 图删掉一些离群细胞 从图中可以看到,n_genes_by_counts > 8000的点更可能是离群点,删除掉
mus = mus[mus.obs.n_genes_by_counts < np.quantile(mus.obs.n_genes_by_counts.values, 0.99), :] # 这里删掉的是最大的1%的细胞
mus = mus[mus.obs.pct_counts_mt < 1, :]
# 标准化counts,结合n_gene_by_counts 图, 使用1e4的来标准化
sc.pp.normalize_total(mus, target_sum=1e4)
# log(x+1),放大差异 默认自然对数
sc.pp.log1p(mus)
# 筛选高可变基因 HVG 使用默认参数,需要log变换
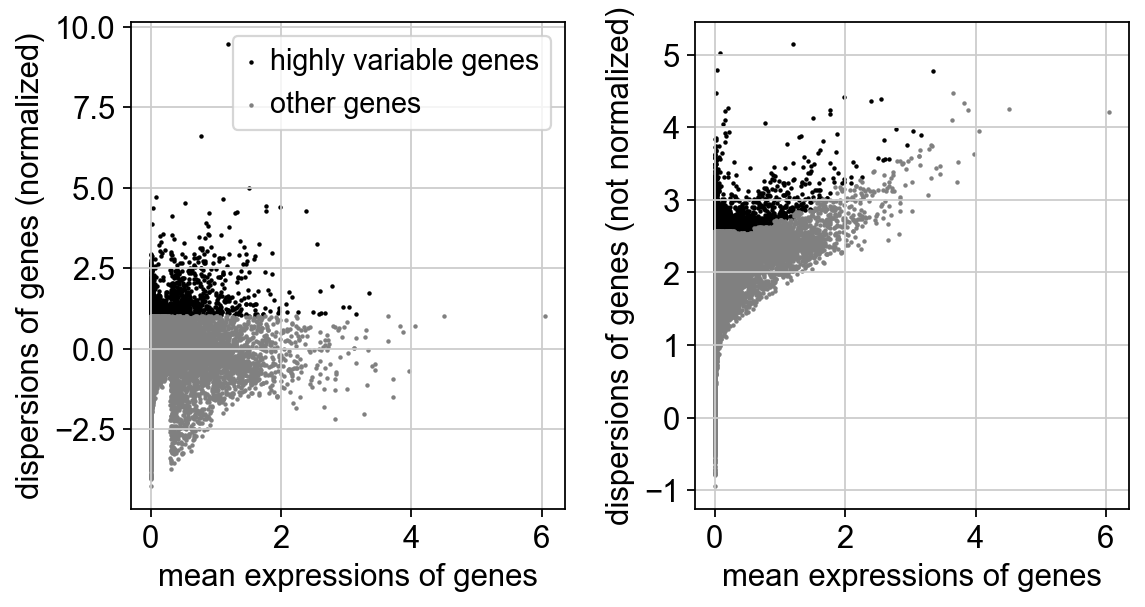
sc.pp.highly_variable_genes(mus, n_top_genes = 2000,min_mean=0.0125, max_mean=3, min_disp=0.5,n_bins=20)
sc.pl.highly_variable_genes(mus)
print(mus.var['highly_variable'].sum())
mus = mus[:, mus.var.highly_variable]
sc.pp.regress_out(mus, ['total_counts', 'pct_counts_mt'])
# 基因方差归一化. 过滤掉标准差大于10的基因
sc.pp.scale(mus, max_value=10)
这里的主要操作有,根据前面的两个阈值筛选细胞,即删掉表达基因数目过高的细胞(可能是barcode中包含多个cell的情形)和线粒体基因表达比例过高的细胞。。之后将所有细胞的基因表达标准化,即每个细胞的所有基因表达总和统一缩放到1e4,这个数值选取基本凭感觉,感觉不会影响特别大。
之后进行$log(x+1)$的表换,$log$变换会影响数据的分布,有生物统计的论文专门讨论该变换会怎样改善后续的数据分析结果。
之后进行高可变基因(HVG)的筛选,HVG是对每个基因进行单特征筛选。对于里面的参数,不清楚的话就用默认参数。
之后是使用线性回归去除协变量’total_counts, pct_counts_mt’的影响,最后是对基因做z-transform,均值为零,方差为1。
注意,以上数据变换,需要根据数据分析结果来调整,没有一个普适的最优流程。例如z-transform,虽然突出了基因的分布差异,但同时也消除了基因的客观表达量差异,因此是否做z-ransform,取决于后面分析的结果输出
normalizing counts per cell
finished (0:00:00)
d:\Anaconda\envs\scanpy\lib\site-packages\scanpy\preprocessing\_normalization.py:170: UserWarning: Received a view of an AnnData. Making a copy.
view_to_actual(adata)
If you pass `n_top_genes`, all cutoffs are ignored.
extracting highly variable genes
finished (0:00:00)
2000
regressing out ['total_counts', 'pct_counts_mt']
sparse input is densified and may lead to high memory use
finished (0:00:18)
完成上述操作后,数据的维度从32285降低到2000,后续的分析将基于这2000HVG进行。
数据降维,聚类
PCA
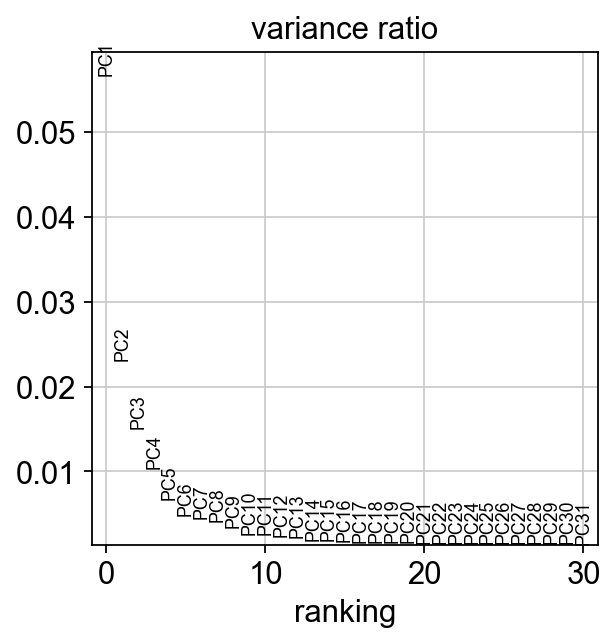
scRNA-seq 数据通常使用PCA降维,之后用UMAP投影到二维做可视化,并利用PCA降维后的数据去构建KNN图并基于图做聚类。一个有争议的点是,通常PCA降维解释的方差比例并不高,例如下面50个主成分仅解释了不到20%的方差,那么这么小的信息量是否足够支持后续分析?
对于这个点,10X的一位工程师给我的回答是,“那么基于PCA分析的结果怎么样呢?”OK,不管从可视化效果还是聚类注释的结果来看,基于PCA降维的结果进行后续流程没有什么很糟糕的表现。那么就follow这套流程好了。后面我们也会看一下直接在原始的基因表达数据上UMAP可视化会有怎样的结果。
sc.tl.pca(mus, svd_solver='arpack')
print('top 5 PCs explained variance ' + str(mus.uns['pca']['variance'][:5]))
print('top 50 PCs explained variance ratio ' + str(mus.uns['pca']['variance_ratio'].sum()))
sc.pl.pca_variance_ratio(mus, log=False)
输出
computing PCA
on highly variable genes
with n_comps=50
finished (0:00:03)
top 5 PCs explained variance [70.06387 28.58396 18.580736 12.756718 8.350449]
top 50 PCs explained variance ratio 0.1930819
# 可视化gene在主成分PC中的载荷(loadings), pca_load_rank的代码在博客的最后
utils.pca_load_rank(mus)
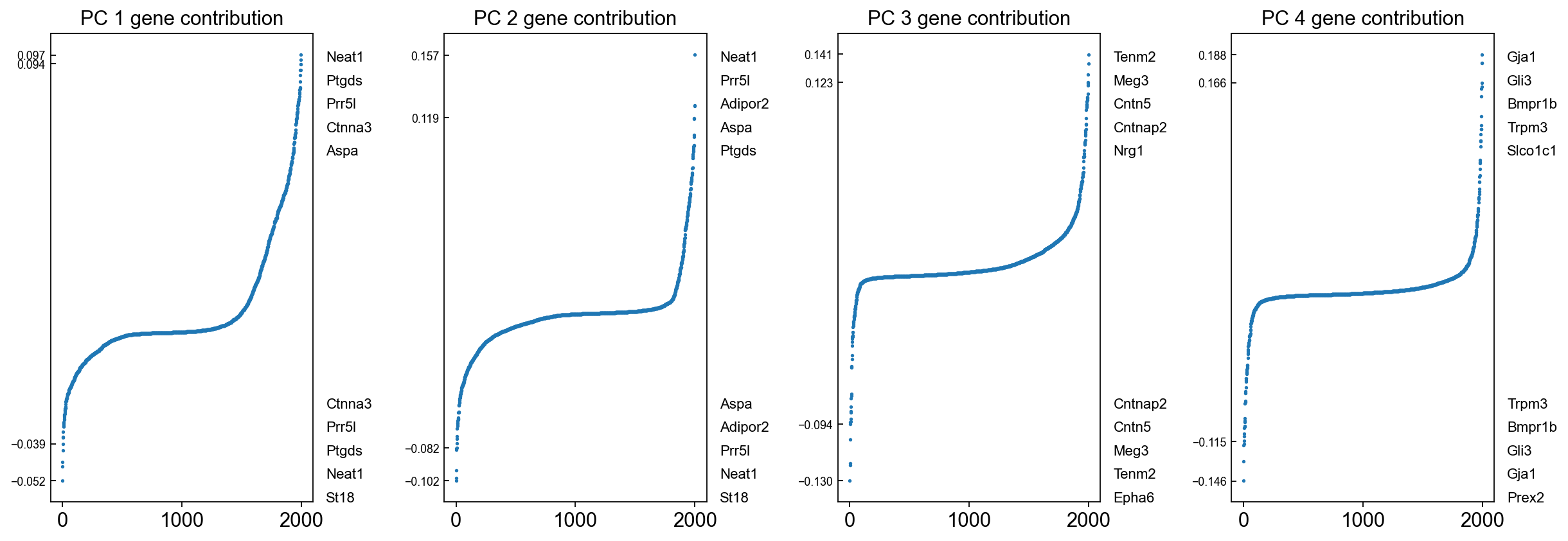
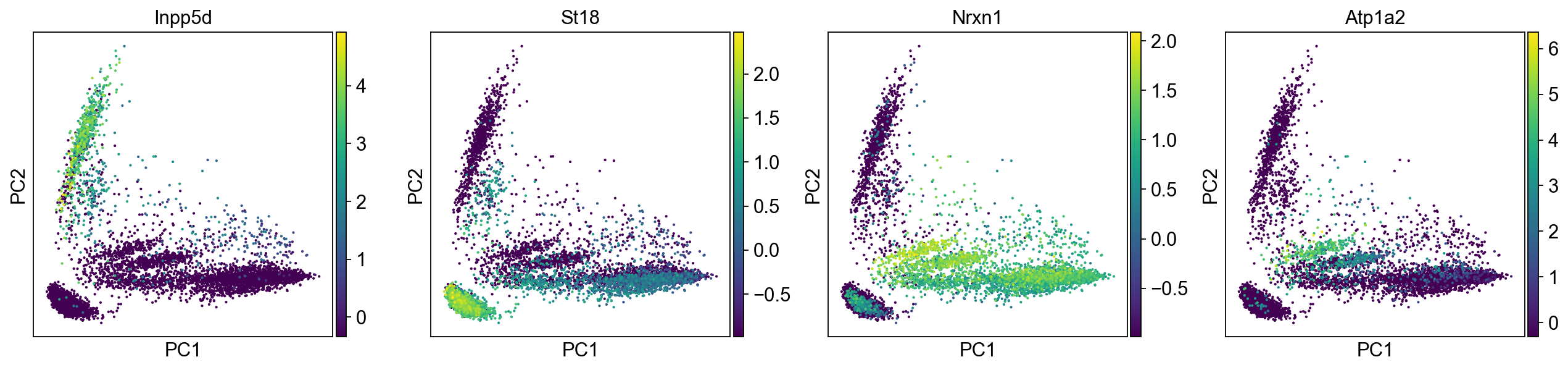
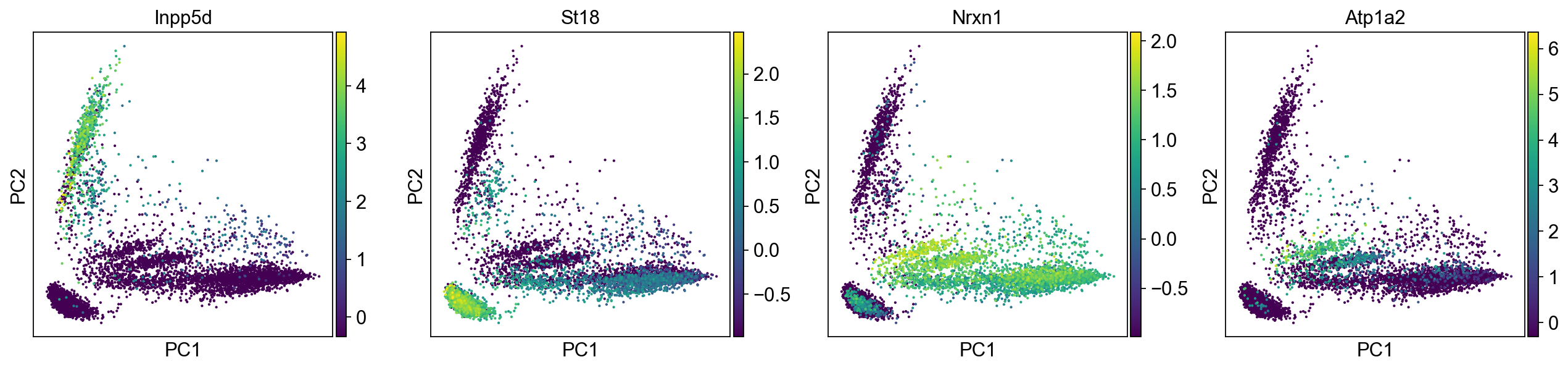
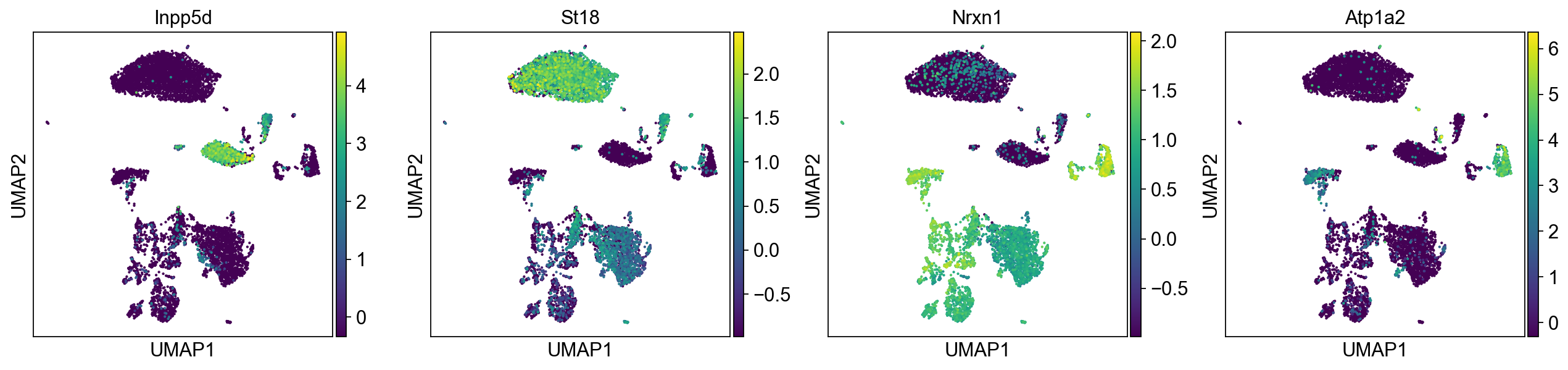
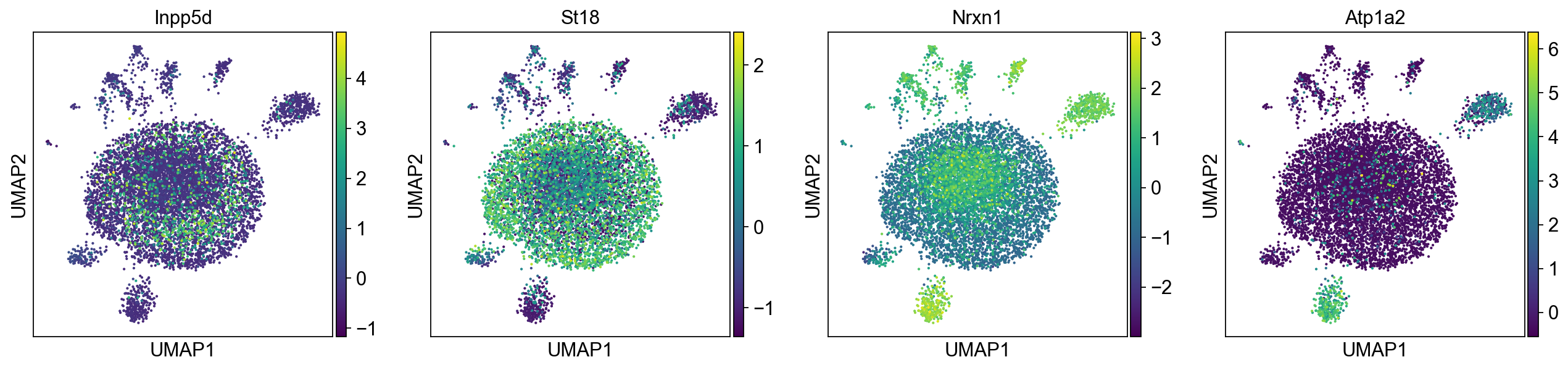
下图展示了用PCA将细胞投影到二维空间后一些基因的表达情况,这里基因选择的是在各个主成分中载荷最大的。建议在完成细胞注释后再回来可视化一下marker gene的表达,看一下各个细胞类型在2维PCA空间中是否有比较明显的模式。
sc.pl.pca(mus, color=['Inpp5d','St18','Nrxn1','Atp1a2'])
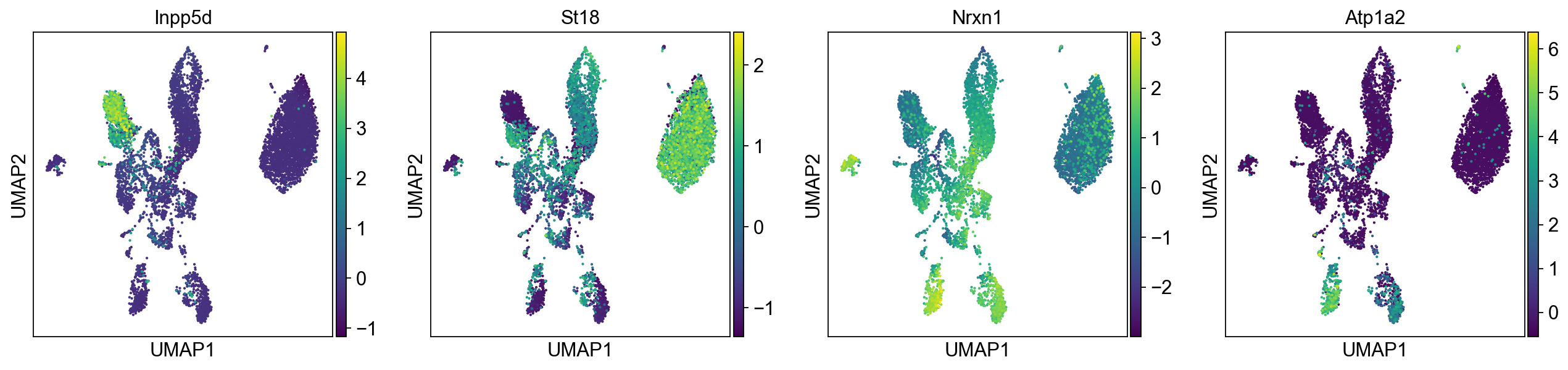
UMAP 可视化,leiden 聚类
首先构造K近邻图,之后UMAP将数据投影到二维空间
sc.pp.neighbors(mus, n_neighbors=10, n_pcs=50)
sc.tl.umap(mus)
# 这里 gene_list 可以先使用PCA 中loading 较大的基因
gene_list = ['Inpp5d','St18','Nrxn1','Atp1a2']
sc.pl.umap(mus, color=gene_list)
输出
computing neighbors
using 'X_pca' with n_pcs = 50
finished (0:00:16)
computing UMAP
finished (0:00:11)
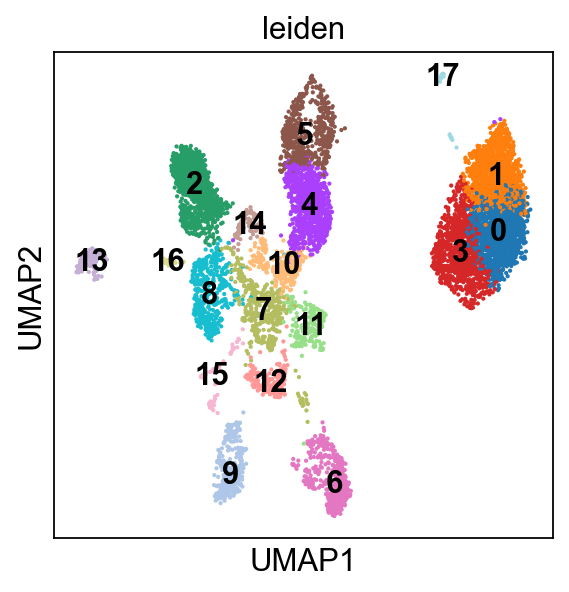
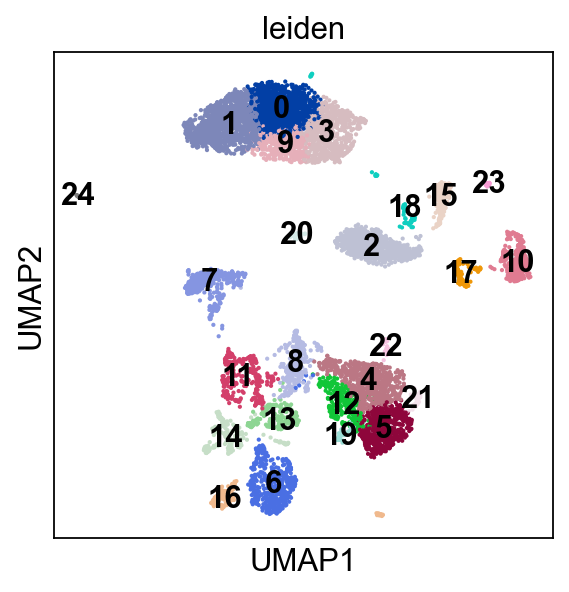
之后使用leiden算法对knn图进行社区划分,leiden算法的超参为resolution, 越大划分的精细度(粒度)越高。怎样确定聚类的粒度,或者说怎样确定聚类的cluster数目是个很困难的问题。结合我看过的一些CNS上的文章,在不同粒度下进行聚类,之后用层次聚类的方法将各个粒度的聚类结果整合起来是一个比较常用的方法,这篇博客中不涉及这个主题。
sc.tl.leiden(mus, resolution = 1,random_state=0)
sc.pl.umap(mus, color=['leiden'],legend_loc='on data')
cluster_num = mus.obs['leiden'].nunique()
cluster_cell_num = []
for i in range(cluster_num):
cluster_cell_num.append((mus.obs['leiden'].values == str(i)).sum())
print('cluster cell number ' + '\n')
print(cluster_cell_num)
输出
cluster cell number
[1272, 1102, 1047, 919, 871, 547, 467, 413, 411, 339, 261, 199, 174, 111, 103, 63, 35, 23]
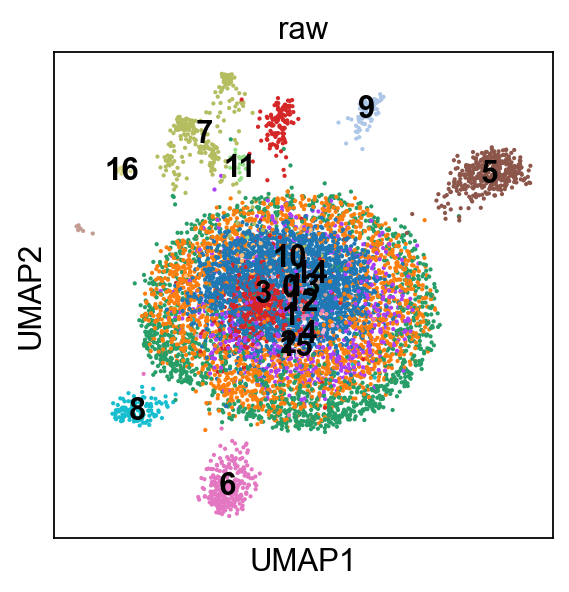
至此,我们将数据划分成18个cluster,之后将讨论怎样将这些cluster注释成有生物学意义的细胞类型。
cluster 注释
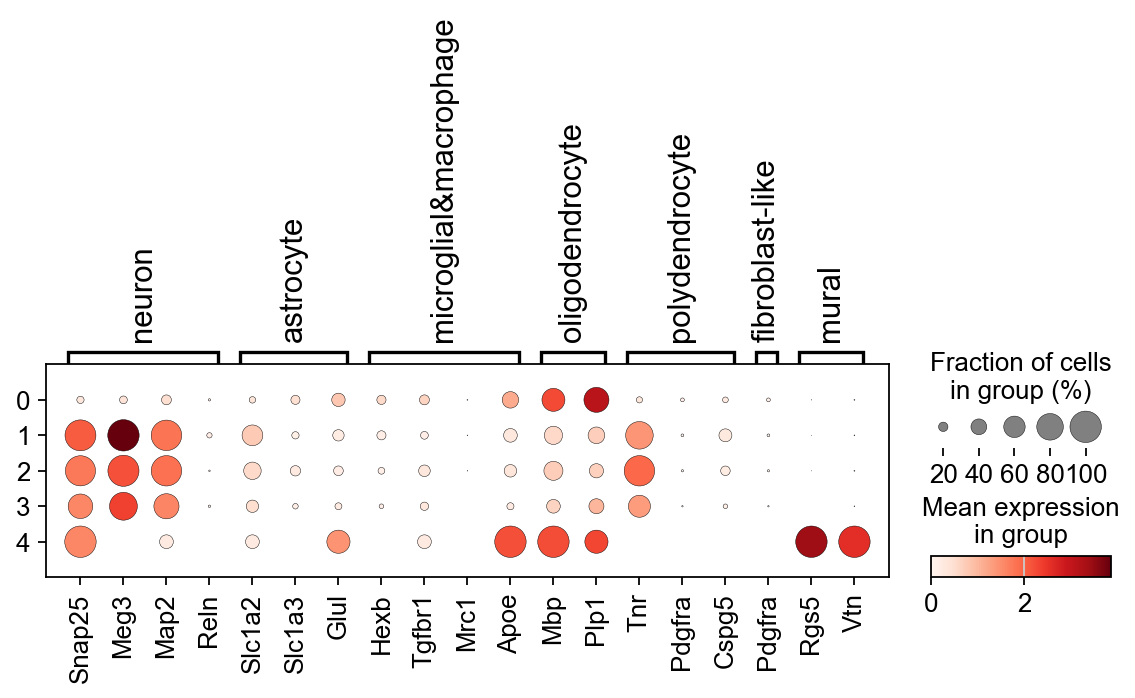
cluster注释通常有两种策略,一种是传统的基于marker gene的注释方法,另一种是有一组已经注释好的数据,可以通过监督学习的方法注释自己的数据。但这种方法的效果受制于注释数据的质量,同时不同的数据之间又可能存在批次效应,因此有时候还是需要用最传统的marker gene的方法进行注释。下面我们将说明怎样使用marker gene 进行注释。
marker gene注释的基本方法就是基于dotplot的可视化效果,去看各个marker gene在各个cluster中的表达情况,如果有特异性表达就可以很容易的完成注释。marker gene注释主要需要考虑三点:
- 有几种潜在的细胞类型
- 这些细胞类型对应的marker有哪些
- 怎样保证注释结果的可靠性
原因如下
- 一般通过统计方法找到的marker gene,很难保证这些marker的生物学意义,例如是否有比较权威的研究发现过这些marker?如果没有的话,生物研究者会质疑。
- 有时一个marker gene 可能会对应多个细胞类型,这时怎么确定注释成哪一个细胞类型。例如neuron下面根据神经递质可以分成十几个亚型,总会有在所有neuron中几乎都是高表达的基因,如Snap25。这时候如果有预先猜测潜在的细胞类型,自然会先用Snap25区分neuron 和 non-neuron,之后对neuron做精细聚类。
- 由于前面的分析是使用的HVG,在筛选过程中,可能有些有意义的marker会被扔掉,这时仅根据HVG中的marker gene是无法有效注释的,需要我们确定好一些有价值的marker,然后在未筛选HVG的数据集上做分析。
对于注释中需要注意的三点,我的建议是,潜在细胞类型通过权威研究结果确定,比如有篇cell和你分析的数据来自同一个组织,那么就用这篇cell中的细胞注释结果为基准进行注释。细胞类型对应的marker,可以从CellMarker数据库中找到,也可以从一些研究论文中去找他们注释用的marker。注释结果可靠性,一方面是marker特异性表达的效果,例如dotplot有非常明显的pattern,另一方面则是marker的可信度,例如cell中研究的marker就比水刊用的marker更可信一些。
下面展示我的注释流程。
raw_mus是最开始的数据,对其做一样的预处理,但不做z-transform。将mus聚类的结果分配到raw_mus上
raw_mus.var_names_make_unique()
utils.stage_1_QC(raw_mus)
raw_mus = raw_mus[raw_mus.obs.n_genes_by_counts < np.quantile(raw_mus.obs.n_genes_by_counts.values, 0.99), :]
raw_mus = raw_mus[raw_mus.obs.pct_counts_mt < 1, :]
sc.pp.normalize_total(raw_mus, target_sum=1e4)
sc.pp.log1p(raw_mus)
raw_mus.obs['leiden'] =mus.obs['leiden']
之后导入下载的CellMarker数据库的小鼠Marker文件,从中选出预先确定好的细胞类型对应的marker,小鼠脑区猜测的细胞类型记录了在cell_list中。绘制dotplot
cell_list = ['Astrocyte','Neuron','Microglial cell','Macrophage', 'Oligodendrocyte',
'Polydendrocyte','Endothelial cell','Fibroblast','Mural cell','Choroid plexus cell','Ependymal cell']
marker_dic, brain_marker = utils.get_marker_dic(cell_list, raw_mus)
sc.pl.dotplot(raw_mus, marker_dic, 'leiden')
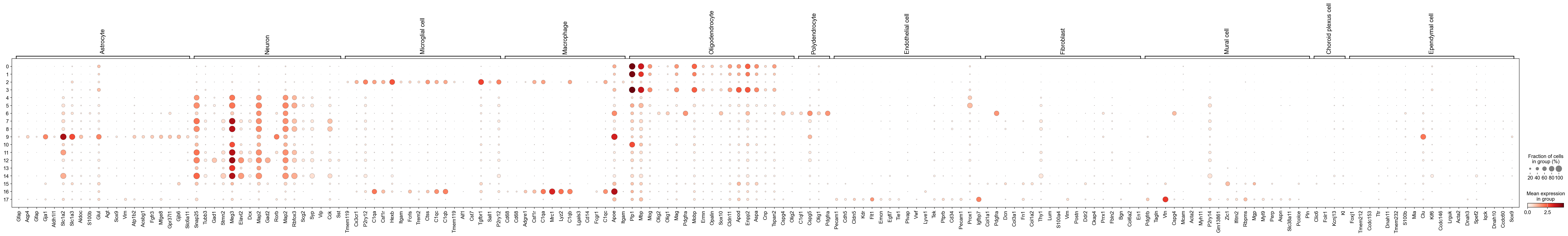
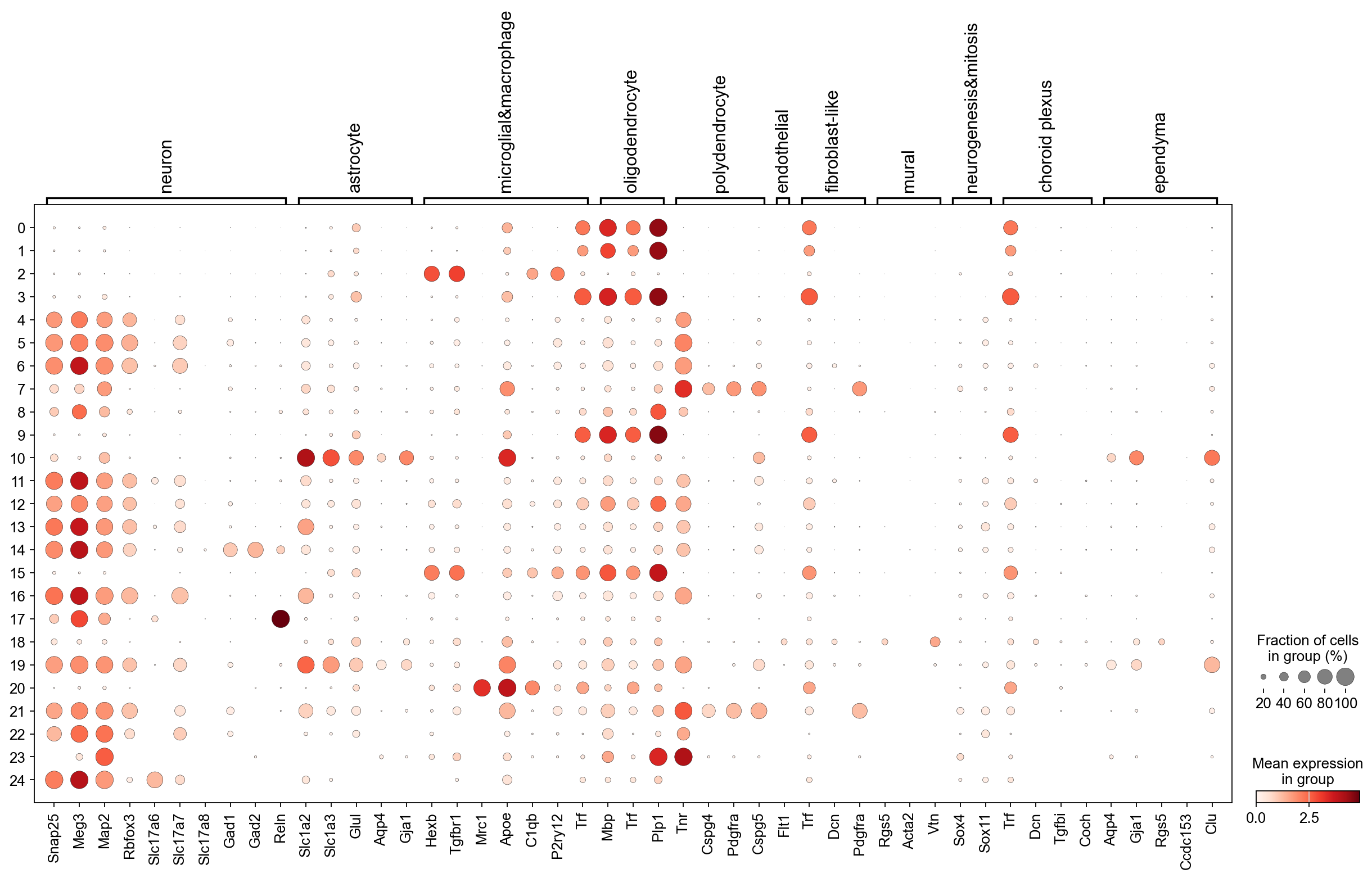
上面的dotplot是非常稀疏的,因此后面要手动选择一些有意义的marker去调整可视化效果
marker_dic = {}
marker_dic['neuron'] = ['snap25','meg3','map2']+ ['reln']
marker_dic['astrocyte'] = ['Slc1a2','slc1a3','glul']
marker_dic['microglial¯ophage'] = ['hexb','tgfbr1','mrc1','apoe']
marker_dic['oligodendrocyte'] = ['mbp','plp1']
marker_dic['polydendrocyte'] = ['tnr','pdgfra','cspg5']
#marker_dic['endothelial'] = ['lgfbp7'] + ['flt1']
marker_dic['fibroblast-like'] = ['lgfbpl1','pdgfra']
marker_dic['mural'] = ['rgs5'] + ['vtn']
#marker_dic['neurogenesis&mitosis'] = ['sox4','sox11']
#marker_dic['choroid plexus'] = ['trf']
#marker_dic['ependyma'] = ['rgs5']
marker_dic = utils.filter_marker(marker_dic, raw_mus)
sc.pl.dotplot(raw_mus, marker_dic, 'leiden')
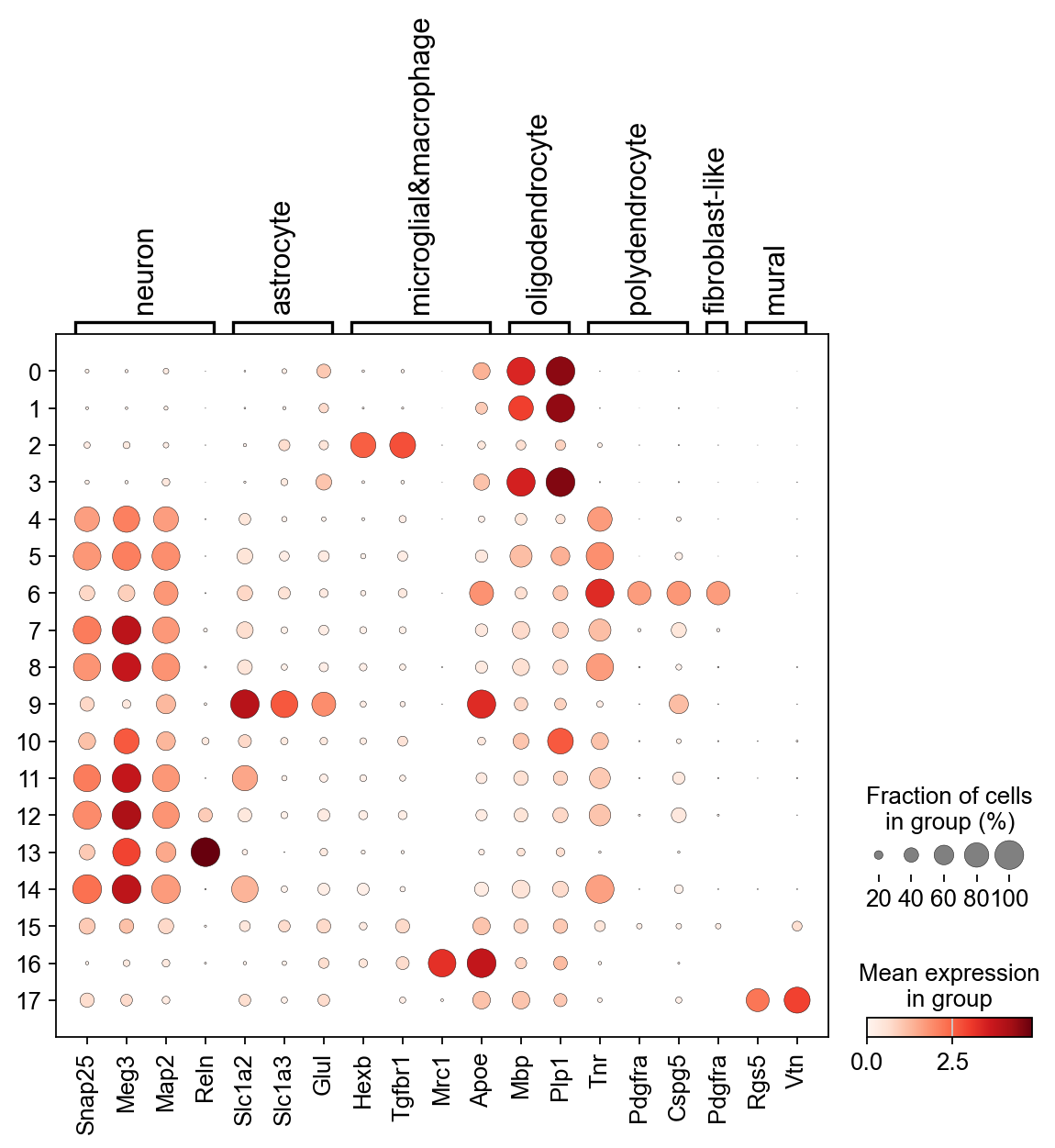
至此,我们得到了一个有着明显pattern的dotplot,并可以进行后续的细胞类型注释。
cell_ann = {}
for i,cluster in enumerate([0,1,3]):
cell_ann[cluster] = 'oligodendrocyte_' + str(i)
for i,cluster in enumerate([4,5,7,8,10,11,12,13,14]):
cell_ann[cluster] = 'neuron_' + str(i)
for i,cluster in enumerate([2,16]):
cell_ann[cluster] = 'microglial¯ophage_' + str(i)
cell_ann[6] = 'polygendrocyte'
cell_ann[9] = 'astrocyte'
cell_ann[17] = 'mural'
cell_ann[15] = 'unknown'
# 这一步是将利用CellMarker的资源,将各个marker的来源(论文期刊)记录下来,看一下marker的可靠性
utils.save_marker(marker_dic, brain_marker, "mhm_marker.txt")
import matplotlib.pyplot as plt
umap_loc = mus.obsm['X_umap']
plt.figure(figsize = (10,10))
color_map = []
for _ in mus.obs['leiden'].values:
color_map.append(mus.uns['leiden_colors'][int(_)])
cluster_label = mus.obs['leiden'].values
legend_loc = []
for i in range(18):
x = umap_loc[(cluster_label == str(i)),0]
y = umap_loc[(cluster_label == str(i)),1]
legend_loc.append((x.mean(),y.mean()))
plt.scatter(x,y,s=0.8,label= str(i) + ' ' + cell_ann[i]+' (' + str(x.shape[0]) + ')', c = mus.uns['leiden_colors'][i])
plt.legend(loc = (1,0.35),markerscale = 5)
plt.title('MHM scRNA-Seq annotation')
legend_loc[-1] = (9,0)
for i,loc in enumerate(legend_loc):
plt.text(loc[0],loc[1],str(i))
#plt.savefig(r'D:\sly_data\figures\MHM_cell_ann.png',bbox_inches = 'tight')
plt.show()
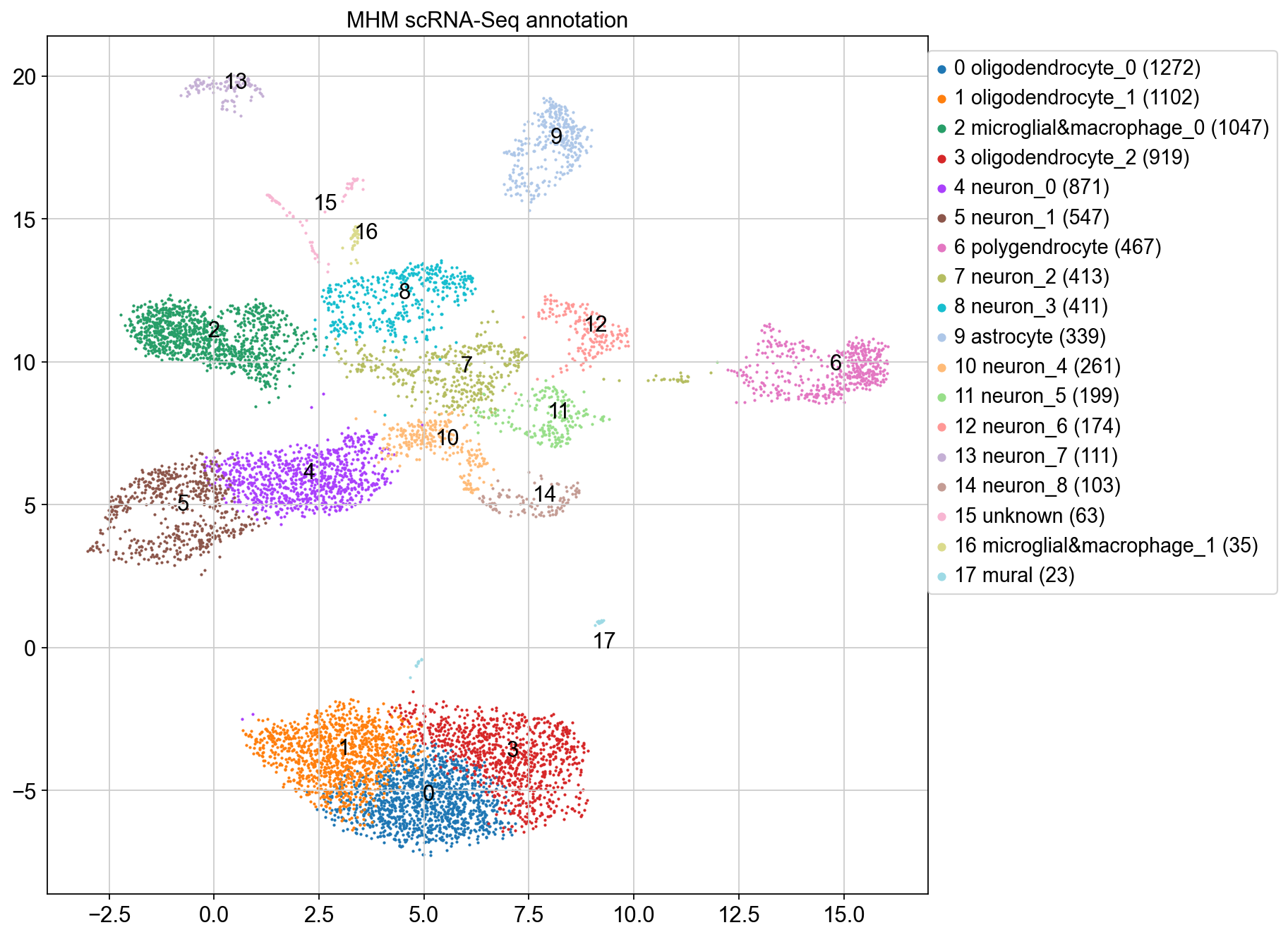
至此我们得到了一个完整的数据集注释结果。
scRNA-seq 的一些问题
这里尝试回答一下导师提出的问题,为什么用PCA+UMAP而不是直接做UMAP,本篇博客暂时不进行理论层面的分析,直接上实验
不使用HVG的分析流程
可以看到,在不进行HVG筛选的情况下,基于PCA的UMAP还是有明显Pattern的,直接做UMAP的就是惨不忍睹了。
umap based on PCA
computing PCA
with n_comps=50
finished (0:00:25)
top 5 PCs explained variance [501.63788 138.9213 92.311386 66.482475 56.421654]
top 50 PCs explained variance ratio 0.08693491

umap based on all genes
下面是直接使用所有基因进行UMAP的代码和结果,代码的主要修改是在scanpy函数中注明,使用哪个key对应的数据做分析。
sc.pp.neighbors(mus, n_neighbors=10,use_rep='X',key_added = 'raw')
sc.tl.umap(mus,neighbors_key='raw')
# 这里 gene_list 可以先使用PCA 中loading 较大的基因
gene_list = ['Inpp5d','St18','Nrxn1','Atp1a2']
sc.pl.umap(mus, color=gene_list)
sc.tl.leiden(mus, resolution = 1,random_state=0,neighbors_key='raw',key_added = 'raw')
sc.pl.umap(mus, color=['raw'],legend_loc='on data')
raw_mus.obs['leiden'] =mus.obs['raw']
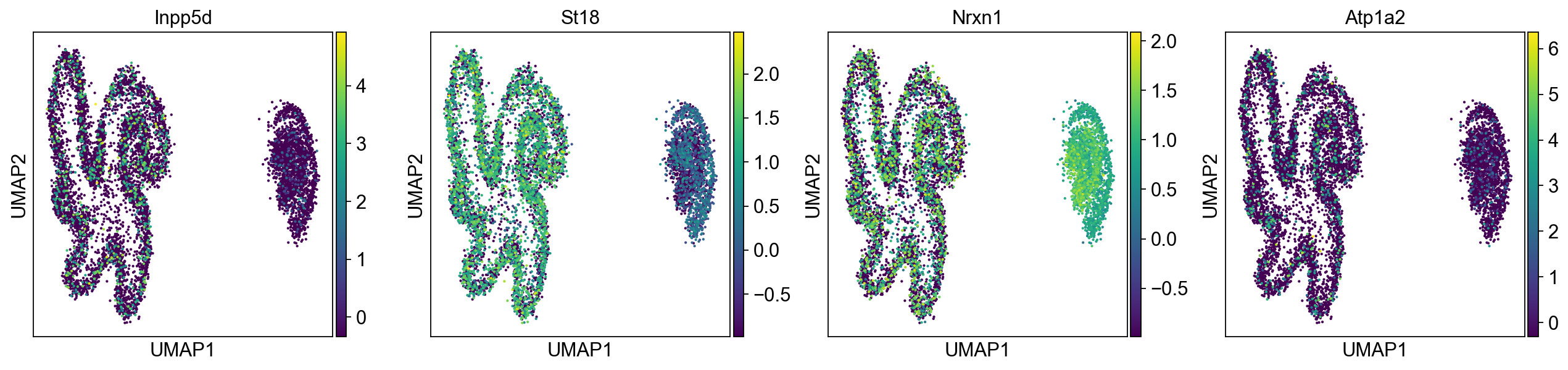
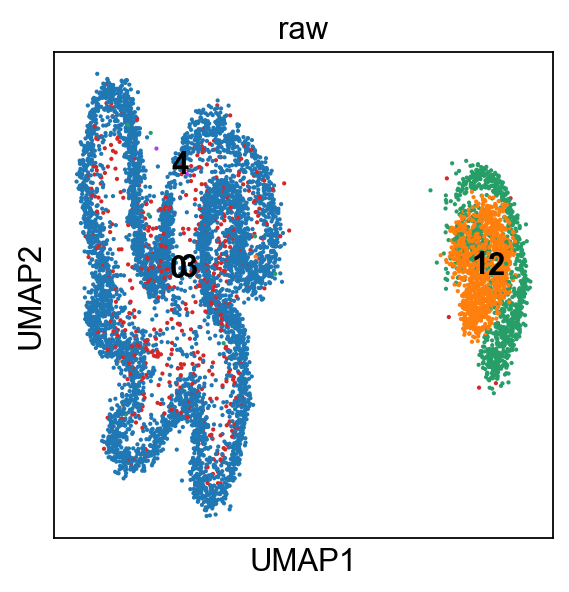
直接做UMAP基本上啥也看不出来。
使用HVG的分析流程
再看一下使用HVG分析的效果。
umap based on PCA of HVG
见scRNA-seq基本分析流程中的结果
umap directly on all HVG
直接做UMAP几乎没有什么好的结果。
总结
最后,简单总结下scRNA-seq的分析流程:
- QC
- HVG筛选,数据变换
- PCA降维
- UMAP可视化,聚类
- 细胞类型注释
数据分析,结果是最重要的。在数据处理逻辑合理的前提下,要从结果出发去考虑问题,去恰当的美化可视化结果。不要一味地尝试一些数学上fancy的东西。毕竟,做的是数据分析,不是数学。
utils 中的一些函数
下面是自己分析中用到的一些可视化函数脚本,供参考
#### plot pca loadings of different PCs, show gene contributions
def pca_load_rank(adata):
hvg_name = adata.var['highly_variable'].index
loadings = adata.varm['PCs']
sort_loadings = np.sort(loadings, axis =0)
sort_ids = np.argsort(loadings, axis = 0)
fig, ax = plt.subplots(nrows = 1, ncols = 4, figsize=(18, 6))
plt.subplots_adjust(wspace =0.5)
for _ in range(4):
pc_load = sort_loadings[:,_]
pc_ids = hvg_name[sort_ids[:,_]]
y_ticks = np.around(np.concatenate((pc_load[[0,4]],pc_load[[-4,-1]])),3)
ax[_].tick_params(axis='y',labelsize=8,direction='in')
ax[_].set_yticks(y_ticks)
ax[_].scatter(x = np.arange(loadings.shape[0]),y = pc_load, s = 2)
for i in range(5):
note = pc_ids[i]
ax[_].text(1.05,0.05*i,s = note,transform=ax[_].transAxes,fontsize=10)
for i in range(1,6):
note = pc_ids[i]
ax[_].text(1.05,0.99-0.05*i,s = note,transform=ax[_].transAxes,fontsize = 10)
ax[_].set_title('PC ' + str(_+1) + ' gene contribution')
ax[_].grid(False)
plt.show()
#### get mus brain marker dictionary
#### 根据cellmarker数据库中提供的marker gene文件,返回marker_gene_dic 和 marker gene dataframe
#### cell_list 为需要整理marker的细胞类型, 需要与marker gene dataframe 中 cell_name 对应
#### raw_adata 为需要注释的单细胞数据, 包含该数据中测得的全部gene
def get_marker_dic(cell_list, raw_adata):
marker = pd.read_excel(r'marker file\Cell_marker_Mouse.xlsx')
brain_marker = marker.loc[marker['tissue_class']=='Brain',:]
print('mouse brain marker number is ' + str(brain_marker.shape[0]))
brain_marker = marker.loc[marker['cell_type']!='Cancer cell',:]
print('mouse brain normal cell marker number is ' + str(brain_marker.shape[0]))
# 这里 cell_list 需要手动输入
cell_list = ['Astrocyte','Neuron','Microglial cell','Macrophage', 'Oligodendrocyte',
'Polydendrocyte','Endothelial cell','Fibroblast','Mural cell','Choroid plexus cell','Ependymal cell']
# 这里导入各个cell type 对应的marker, 对于 marker 数目较多的细胞类型,我们使用文献中记录次数top20的marker
marker_dic = {}
for cell in cell_list:
tmp_marker = brain_marker.loc[brain_marker.loc[:,'cell_name'] == cell ,:]
if tmp_marker.loc[:,'marker'].value_counts().shape[0] > 20:
tmp_marker_list = tmp_marker.loc[:,'marker'].value_counts().index[:20]
else:
tmp_marker_list = np.unique(tmp_marker.loc[:,'marker'].values)
marker_dic[cell] = tmp_marker_list
# 选择在scRNA seq 数据中有记录的 marker gene
marker_dic = filter_marker(marker_dic, raw_adata)
return marker_dic, brain_marker
#### select gene in both marker_dic and raw_adata gene_ids
# 有时获得的marker_dic中的基因没有全部包含在raw_adata的gene_ids中,需要过滤
def filter_marker(marker_dic, raw_adata):
# 选择在scRNA seq 数据中有记录的 marker gene
ori_id = raw_adata.var['gene_ids'].index.values.copy()
cap_id = raw_adata.var['gene_ids'].index.values.copy()
for i,_ in enumerate(cap_id):
cap_id[i] = _.upper()
for key in marker_dic.keys():
tmp = marker_dic[key]
new_tmp = []
for i,_ in enumerate(tmp):
if tmp[i].upper() in cap_id:
new_tmp.append(ori_id[cap_id==tmp[i].upper()][0])
marker_dic[key] = new_tmp
return marker_dic
#### save marker and its source in txt file
def save_marker(marker_dic, brain_marker, path):
marker_list = []
for key in marker_dic.keys():
marker_list += marker_dic[key]
# 打开文件,如果文件不存在则创建新文件
with open(path, "w") as file:
file.write('marker name' + '\t' + 'journal' + '\n')
for marker in marker_list:
marker_meta = brain_marker.loc[brain_marker.loc[:,'marker'] == marker,:]
file.write(marker + "\t" )
for _ in marker_meta.loc[:,'journal'].unique():
file.write(_ + "\t")
file.write("\n")
print("数据已成功逐行写入文件!")
return None