本篇博客主要记录自己最近在使用10X的cellranger系列软件(cellranger, cellranger-atac, cellranger-arc)过程中遇到的一个问题及解决过程,顺带整理下相关的软件使用步骤,方便后续工作查找。我们首先从bug说起。
BUG修复
问题概述
遇到的问题是,手头一批10X-multiome数据在使用cellranger-arc count 进行处理时出现了报错,初始报错如下
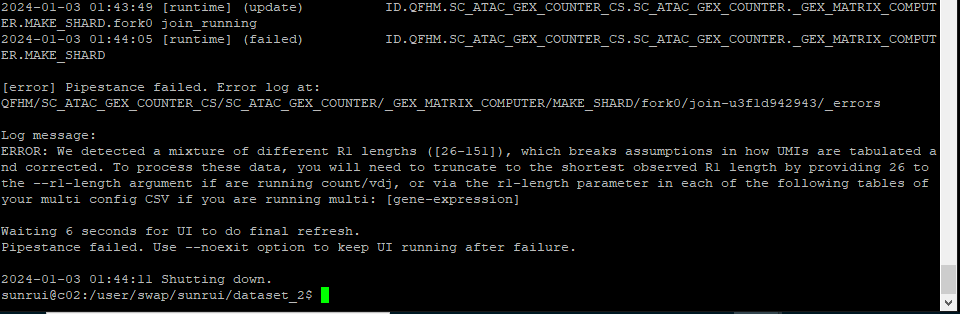
和合作方测序工程师沟通后,反馈结果如下
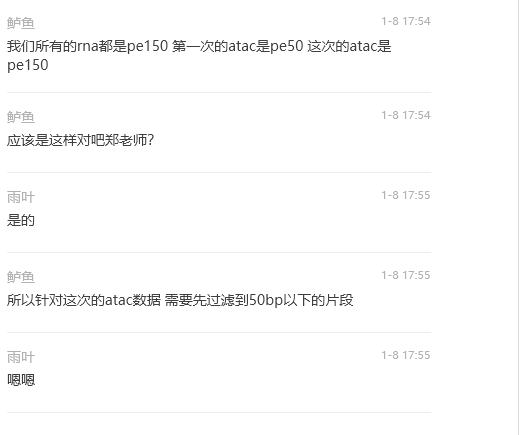
结合上面报错,看起来问题是在测序时使用了更长的测序深度,导致出现长度不一致的片段。
问题解决办法
2024.01.31 补充:
10X的rawdata一般是没有问题的,测序公司通常会将rawdata做一次QC得到cleandata,在这个过程中会对片段进行一些过滤或者裁剪,所以得到的cleandata并不是rawdata的一个子集,裁剪的过程中可能会导致片段的长度不一,因此使用cellranger处理rawdata就可以规避这个问题。淦!
先说明怎样解决这个,后面再记录我确定这个bug的过程。
问题的原因是这批数据的RNA-seq数据中,双端测序数据R1,R2中出现了两种长度的片段,26和151,所以只要把片段长度处理成一致即可,或者截断到26或者把26的片段都过滤掉。最后解决用的是把26片段全部过滤,应用fastp过滤掉长度不等于151的片段,命令如下
(base) sunrui@n06:/work/swap/sunrui/rna_seq/clean_rna/CY/CY-HM-G$ fastp -i CYHM_S1_L001_R1_001.fastq.gz \
-I CYHM_S1_L001_R2_001.fastq.gz \
-o /work/swap/sunrui/rna_seq/clean_rna/CY/fCY-HM-G/fCYHM_S1_L001_R1_001.fastq.gz \
-O /work/swap/sunrui/rna_seq/clean_rna/CY/fCY-HM-G/fCYHM_S1_L001_R2_001.fastq.gz \
--length_required=151 \
--thread=16
Read1 before filtering:
total reads: 453120558
total bases: 68336384444
Q20 bases: 53504228535(78.2954%)
Q30 bases: 45313963764(66.3102%)
Read2 before filtering:
total reads: 453120558
total bases: 68342512478
Q20 bases: 66114787475(96.7404%)
Q30 bases: 62366018303(91.2551%)
Read1 after filtering:
total reads: 449806960
total bases: 67920823659
Q20 bases: 53176824991(78.2924%)
Q30 bases: 45031841437(66.3005%)
Read2 after filtering:
total reads: 449806960
total bases: 67920823659
Q20 bases: 65749637730(96.8034%)
Q30 bases: 62042156342(91.3448%)
Filtering result:
reads passed filter: 899613920
reads failed due to low quality: 2088
reads failed due to too many N: 0
reads failed due to too short: 6625108
reads with adapter trimmed: 0
bases trimmed due to adapters: 0
Duplication rate: 6.15658%
Insert size peak (evaluated by paired-end reads): 271
JSON report: fastp.json
HTML report: fastp.html
fastp -i CYHM_S1_L001_R1_001.fastq.gz -I CYHM_S1_L001_R2_001.fastq.gz -o /work/swap/sunrui/rna_seq/clean_rna/CY/fCY-HM-G/fCYHM_S1_L001_R1_001.fastq.gz -O /work/swap/sunrui/rna_seq/clean_rna/CY/fCY-HM-G/fCYHM_S1_L001_R2_001.fastq.gz --length_required=150 --thread=16
fastp v0.23.4, time used: 1959 seconds
之后运行cellranger-arc count, 问题成功解决!!!
- ATAC peak locations: /work/swap/sunrui/rna_seq/clean_rna/CY/CYHM/outs/atac_peaks.bed
- ATAC smoothed transposition site track: /work/swap/sunrui/rna_seq/clean_rna/CY/CYHM/outs/atac_cut_sites.bigwig
- ATAC peak annotations based on proximal genes: /work/swap/sunrui/rna_seq/clean_rna/CY/CYHM/outs/atac_peak_annotation.tsv
Waiting 6 seconds for UI to do final refresh.
Pipestance completed successfully!
2024-01-20 23:32:21 Shutting down.
问题解决流程
这里记录下我是怎样将问题逐步定位到RNA-seq数据中片段长度不一致的。
初次解决尝试
直接参考图1中10X的日志信息,在cellranger-arc count中设置参数 –r1-length=26, 报错。 原因是cellranger-arc count中根本没有这个参数,致信10X官方后也得到了这个回复,确实没有。淦!
之后考虑分别跑rna-seq和atac-seq,10X 官网上倒是有提怎么把多组学数据分别用RNA和ATAC的流程处理,但是这样做和直接cellranger-arc count之间的差异有多大,需要后续咨询10X官方。
开始先做的cellranger count看RNA-seq效果,此时需要加额外参数 –r1-length=26 和 –chemistry=”ARC-v1”。最后结果RNA-seq是可行的,能出结果并且结果不太烂。
这里补一个长翼蝠海马的RNA运行结果, 脚本命令和结果如下
#!/bin/sh
#SBATCH --output=CYHM.out
#SBATCH --error=CYHM.err
#SBATCH --mail-type=ALL
#SBATCH --mail-user=sunrui171@mails.ucas.edu.cn
cellranger count --id=CYHM \
--sample=CYHM \
--chemistry='ARC-v1' \
--r1-length=26 \
--transcriptome=/temp/swap/sunrui/genome/mfu \
--fastqs=/work/swap/sunrui/rna_seq/clean_rna/CY/CY-HM-G \
--localcores=48 \
--localmem=64
2024-01-20 01:13:03 [runtime] (ready) ID.CYHM.SC_RNA_COUNTER_CS.SC_MULTI_CORE.MULTI_REPORTER.CHOOSE_CLOUPE
2024-01-20 01:13:03 [runtime] (run:local) ID.CYHM.SC_RNA_COUNTER_CS.SC_MULTI_CORE.MULTI_REPORTER.CHOOSE_CLOUPE .fork0.chnk0.main
2024-01-20 01:13:04 [runtime] (chunks_complete) ID.CYHM.SC_RNA_COUNTER_CS.SC_MULTI_CORE.MULTI_REPORTER.CHOOSE_CLOUPE
2024-01-20 01:13:45 [runtime] (join_complete) ID.CYHM.SC_RNA_COUNTER_CS.SC_MULTI_CORE.MULTI_GEM_WELL_PROCESSOR.COU NT_GEM_WELL_PROCESSOR._BASIC_SC_RNA_COUNTER.WRITE_POS_BAM
Outputs:
- Run summary HTML: /work/swap/sunrui/rna_count/CY/CYHM/outs/web_summary.html
- Run summary CSV: /work/swap/sunrui/rna_count/CY/CYHM/outs/metrics_summary.csv
- BAM: /work/swap/sunrui/rna_count/CY/CYHM/outs/possorted_genome_bam.bam
- BAM BAI index: /work/swap/sunrui/rna_count/CY/CYHM/outs/possorted_genome_bam.bam.bai
- BAM CSI index: null
- Filtered feature-barcode matrices MEX: /work/swap/sunrui/rna_count/CY/CYHM/outs/filtered_feature_bc_matrix
- Filtered feature-barcode matrices HDF5: /work/swap/sunrui/rna_count/CY/CYHM/outs/filtered_feature_bc_matrix.h5
- Unfiltered feature-barcode matrices MEX: /work/swap/sunrui/rna_count/CY/CYHM/outs/raw_feature_bc_matrix
- Unfiltered feature-barcode matrices HDF5: /work/swap/sunrui/rna_count/CY/CYHM/outs/raw_feature_bc_matrix.h5
- Secondary analysis output CSV: /work/swap/sunrui/rna_count/CY/CYHM/outs/analysis
- Per-molecule read information: /work/swap/sunrui/rna_count/CY/CYHM/outs/molecule_info.h5
- CRISPR-specific analysis: null
- Antibody aggregate barcodes: null
- Loupe Browser file: /work/swap/sunrui/rna_count/CY/CYHM/outs/cloupe.cloupe
- Feature Reference: null
- Target Panel File: null
- Probe Set File: null
Waiting 6 seconds for UI to do final refresh.
Pipestance completed successfully!
2024-01-20 01:14:15 Shutting down.
Saving pipestance info to "CYHM/CYHM.mri.tgz"
^Z
[7]+ Stopped tail -f CYHM.out
可以看到在只设置截断r1为26的情形下,RNA是能够正常运行的(cellranger 会自动把r2截断到r1相同长度)。
后续解决尝试
后面决定还是优先考虑用多组学流程去做,毕竟多组学比单组学要贵好多并且质量是要低于单组学的,要是只看RNA,实在是亏死……
2024.01.16
按照报错信息,想直接拿一个工具把测序数据中违规的片段过滤掉。过滤工具尝试的fastp,按照图2中合作方测序工程师的反馈,扔掉片段长度小于50的。(10X报错是26,合作方为啥要扔50的?)
开始只处理了ATAC-seq数据,用的工具是fastp,处理时出现一个问题,fastp貌似只支持R1,R2两条序列同时处理,但ATAC数据有R1,R2,R3,所以没法同时处理,最后决定对每个Read 单独处理,就是
fastp -in R1 -out R1_f --r1-length=50
fastp -in R2 ...... # 这里的命令不是正确的fastp命令,只是一个示意
fastp -in R3 ......
把处理完后的ATAC数据扔到cellranger-arc count上操作,继续报错,结果如下
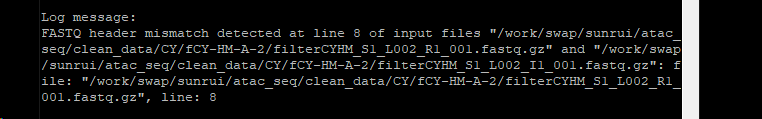
可以看到问题是说过滤后数据的第八行不匹配,用
zcat file | head -n 20
查看R1,R2,R3后发现,有的片段,R1中过滤掉了,但是R2,R3中没有,所以出现的问题。 猜测,要是将一个片段R1,R2,R3同时过滤掉就没有问题了?
后面又问合作方工程师,说让我只用 fastp -in file1 -out file2 这样的默认参数试一下(我很怀疑,这样还是分开处理,怎么解决上面的问题?)
果不其然,还是报错,就是做了个质控,还是遇到不匹配问题……
2024.01.19
这一次开始思考,图一报错中的片段长度不一,到底是RNA引起的还是ATAC引起的还是两者都有,为了确定这一点,将RNA和ATAC数据不做任何处理分别跑cellranger 和 cellranger-arc count。
下面确定RNA-seq测序问题,长翼蝠海马和长翼蝠垂体的报错如下,
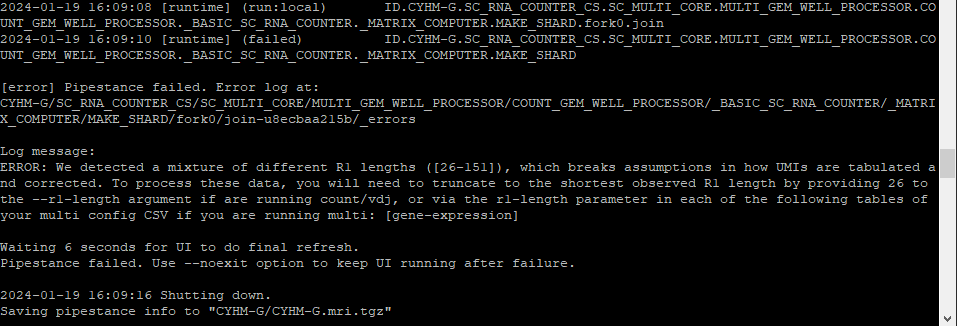
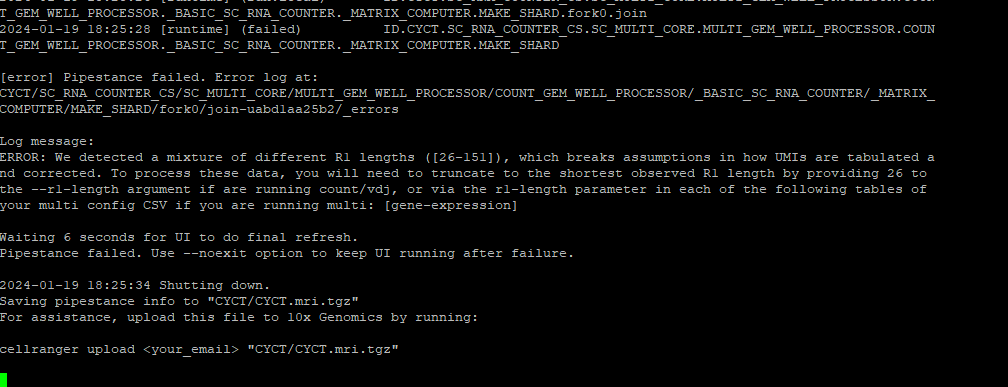
这是长翼蝠垂体RNA-seq处理使用的脚本,可以看到对RNA-seq,不设置–r1-length=26时会出现相同的报错。但是和图1的报错有冲突,图1报错的时候是在ATAC的步骤上出错的。(这里我犯了一个很严重的错误,我忽视了日志保存地址中的*ATAC-GEX*这个信息,先入为主的认为错误实在ATAC步骤里出现的)。
#!/bin/sh
#SBATCH --output=CYCT.out
#SBATCH --error=CYCT.err
#SBATCH --mail-type=ALL
#SBATCH --mail-user=sunrui171@mails.ucas.edu.cn
cellranger count --id=CYCT \
--sample=CYCT \
--chemistry='ARC-v1' \
--transcriptome=/temp/swap/sunrui/genome/mfu \
--fastqs=/work/swap/sunrui/rna_seq/clean_rna/CY/CY-CT-G \
--localcores=48 \
--localmem=64
下面看ATAC-seq数据有没有问题, 报错如下
2024-01-19 17:32:33 [runtime] (chunks_complete) ID.CYHM.SC_ATAC_COUNTER_CS.SC_ATAC_COUNTER._BASIC_SC_ATAC_COUNTER._ATAC_MATRIX_COMPUTER.ALIGN_ATAC_READS
2024-01-19 17:32:33 [runtime] (run:local) ID.CYHM.SC_ATAC_COUNTER_CS.SC_ATAC_COUNTER._BASIC_SC_ATAC_COUNTER._ATAC_MATRIX_COMPUTER.ALIGN_ATAC_READS.fork0.join
2024-01-19 17:32:33 [runtime] (failed) ID.CYHM.SC_ATAC_COUNTER_CS.SC_ATAC_COUNTER._BASIC_SC_ATAC_COUNTER._ATAC_MATRIX_COMPUTER.ALIGN_ATAC_READS
[error] Pipestance failed. Error log at:
CYHM/SC_ATAC_COUNTER_CS/SC_ATAC_COUNTER/_BASIC_SC_ATAC_COUNTER/_ATAC_MATRIX_COMPUTER/ALIGN_ATAC_READS/fork0/join-uae59aa3032/_errors
Log message:
2.2% (< 10%) of read pairs have a valid 10x barcode. This could be a result of poor sequencing quality, a sample mixup, or running the wrong pipeline, for example, running `cellranger-atac` on Multiome ATAC + GEX data, or vice versa.
Waiting 6 seconds for UI to do final refresh.
Pipestance failed. Use --noexit option to keep UI running after failure.
2024-01-19 17:32:39 Shutting down.
运行脚本如下:
(base) sunrui@n07:/work/swap/sunrui/atac_count/CY$ cat CYHM.slurm
#!/bin/bash
#SBATCH --output=CYHM_atac.out
#SBATCH --error=CYHM_atac.err
#SBATCH --mail-type=ALL
#SBATCH --mail-user=sunrui171@mails.ucas.edu.cn
#SBATCH --nodelist=n07
cellranger-atac count --id=CYHM \
--reference=/temp/swap/sunrui/genome/mfu \
--fastqs=/work/swap/sunrui/atac_seq/clean_data/CY/CYHM \
# --sample=mysample \
--localcores=48 \
--localmem=64 \
--chemistry='ARC-v1'
上面的ATAC报错居然没有先报错说ATAC这边Read有问题,而是说有效barcode太少。这种报错我感觉更像是chemistry这个参数没有设置合适导致的。我又看了下cellranger-atac count 的帮助,发现确实没有–chemistry这个参数,但是这个用法10X官网上确提到了,无语,回头必须质问10X,cellranger-atac count 到底是否支持–chemistry的参数设置?
到这一步重新猜测,是不是ATAC是OK的,我只要把RNA处理一下就能在cellranger-arc count上跑了?
截止到目前想到两个需要进一步尝试的策略
- 用截断后的RNA-seq加原始的ATAC-seq走流程
- RNA-seq截断, ATAC也截断, ATAC的截断得自己写脚本啊啊啊啊啊啊啊啊啊啊,这样再不成只能宣布或者只看RNA或者让合作者再找公司解决数据问题了,淦!
尝试解决
fastp 同时处理长翼蝠垂体RNA-seq双端数据,设置的是截断长度为26bp,fastp结果如下(fastp双端运行中途没有任何输出,安心等15min再说)
(base) sunrui@n07:/work/swap/sunrui/rna_seq/clean_rna/CY/CY-CT-G$ fastp -i CYCT_S1_L001_R1_001.fastq.gz -I CYCT_S1_L001_R2_001.fastq.gz -o fCYCT_S1_L001_R1_001.fastq.gz -O fCYCT_S1_L001_R2_001.fastq.gz -b 26 -w 16
Read1 before filtering:
total reads: 406790200
total bases: 61361573650
Q20 bases: 46685950907(76.0834%)
Q30 bases: 37543113863(61.1834%)
Read2 before filtering:
total reads: 406790200
total bases: 61360777858
Q20 bases: 58763295092(95.7669%)
Q30 bases: 54787082253(89.2868%)
Read1 after filtering:
total reads: 404866206
total bases: 10526521033
Q20 bases: 10321364264(98.051%)
Q30 bases: 9909249652(94.136%)
Read2 after filtering:
total reads: 404866206
total bases: 10526486597
Q20 bases: 10292391829(97.7761%)
Q30 bases: 9817541273(93.2651%)
Filtering result:
reads passed filter: 809732412
reads failed due to low quality: 3824906
reads failed due to too many N: 0
reads failed due to too short: 23082
reads with adapter trimmed: 4
bases trimmed due to adapters: 194
Duplication rate: 5.28522%
Insert size peak (evaluated by paired-end reads): 271
JSON report: fastp.json
HTML report: fastp.html
fastp -i CYCT_S1_L001_R1_001.fastq.gz -I CYCT_S1_L001_R2_001.fastq.gz -o fCYCT_S1_L001_R1_001.fastq.gz -O fCYCT_S1_L001_R2_001.fastq.gz -b 26 -w 16
fastp v0.23.4, time used: 679 seconds
应用cellranger-arc count 处理应用fastp 截断 R1,R2的RNA-seq(ATAC未处理),结果如下:
2024-01-19 21:30:45 [runtime] (split_complete) ID.CYCT.SC_ATAC_GEX_COUNTER_CS.SC_ATAC_GEX_COUNTER._GEX_MATRIX_COMPUT ER.MAKE_SHARD
2024-01-19 21:30:45 [runtime] (run:local) ID.CYCT.SC_ATAC_GEX_COUNTER_CS.SC_ATAC_GEX_COUNTER._GEX_MATRIX_COMPUT ER.MAKE_SHARD.fork0.chnk0.main
2024-01-19 21:31:12 [runtime] (failed) ID.CYCT.SC_ATAC_GEX_COUNTER_CS.SC_ATAC_GEX_COUNTER._GEX_MATRIX_COMPUT ER.MAKE_SHARD
[error] Pipestance failed. Error log at:
CYCT/SC_ATAC_GEX_COUNTER_CS/SC_ATAC_GEX_COUNTER/_GEX_MATRIX_COMPUTER/MAKE_SHARD/fork0/chnk0-ub9efaa7985/_errors
Log message:
IO error in FASTQ file '"/work/swap/sunrui/rna_seq/clean_rna/CY/fCY-CT-G/fCYCT_S1_L001_R2_001.fastq.gz"', line: 85795 00: unexpected end of file
Waiting 6 seconds for UI to do final refresh.
Pipestance failed. Use --noexit option to keep UI running after failure.
2024-01-19 21:31:18 Shutting down.
^Z
[2]+ Stopped tail -f CYCT.out
这里发现之前自己一个判断错误,最初的报错是error log 保存在sc_ATAC_GEX文件夹下,但是实际出问题的还是RNA数据,所以只要处理好RNA就够了。之前一直理解错了,尴尬。
看一下出问题的部分
(base) sunrui@n07:/work/swap/sunrui/rna_seq/clean_rna/CY/fCY-CT-G$ zcat fCYCT_S1_L001_R2_001.fastq.gz | sed -n '8579400,857960p'
gzip: fCYCT_S1_L001_R2_001.fastq.gz: unexpected end of file
FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFF:FFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFF,FFFFFFFFFFFFFFFFFFFF:FFFFFFFFFF
这次是.fastq.gz文件格式的问题,试一下按照合作方工程师说的截断到50?再试一下啥也不做?
下面这个实验结果是长翼蝠垂体的多组学数据,用的RNA-seq是fastp截断到51的长度,运行结果如下:
2024-01-20 01:12:36 [runtime] (update) ID.CYCT.SC_ATAC_GEX_COUNTER_CS.SC_ATAC_GEX_COUNTER._GEX_MATRIX_COMPUTR.MAKE_SHARD.fork0 join_running
2024-01-20 01:12:43 [runtime] (failed) ID.CYCT.SC_ATAC_GEX_COUNTER_CS.SC_ATAC_GEX_COUNTER._GEX_MATRIX_COMPUTR.MAKE_SHARD
[error] Pipestance failed. Error log at:
CYCT/SC_ATAC_GEX_COUNTER_CS/SC_ATAC_GEX_COUNTER/_GEX_MATRIX_COMPUTER/MAKE_SHARD/fork0/join-ub9efaa7983/_errors
Log message:
ERROR: We detected a mixture of different R1 lengths ([26-51]), which breaks assumptions in how UMIs are tabulated an corrected. To process these data, you will need to truncate to the shortest observed R1 length by providing 26 to th --r1-length argument if are running count/vdj, or via the r1-length parameter in each of the following tables of you multi config CSV if you are running multi: [gene-expression]
Waiting 6 seconds for UI to do final refresh.
Pipestance failed. Use --noexit option to keep UI running after failure.
2024-01-20 01:12:49 Shutting down.
从这里我们基本确定问题所在了,在RNA数据中,混入了一小部分长度不等于151(长26)的片段,只要把这些长度不等于151的片段过滤掉,程序应该就能正常运行了。
做这个判断的原因是,最初报错是26-151,现在变成了26-51,由于我们用的是截断而不是过滤,所以应该是大量151片段被截断到51,但是原先的26的片段并没有被丢弃,所以依旧长短不一致。而当时直接用cellranger-count –r1-length=26时,所有片段都被截断到26,所以可行。
因此,以后再遇到这种问题时,应当先确定RNA-seq数据中fastq.gz数据中的片段长度是多少,将短的过滤,长的截断,长度统一后再处理。
问题解决
开头说的那样,过滤短RNA片段后再cellranger-arc count.
一些残存问题
下面是自己处理问题过程中想到的几个问题,先记下来等后面解决
- 10X多组学数据分别跑RNA,ATAC会有什么影响?
- 为什么ATAC-seq数据有R1R2R3,不是双端测序吗?
- 是否ATAC-seq数据允许不等长的片段?我统计的ATAC片段好像是长度不一的。
- 提醒10X修正一些报错日志,提醒fastp给个运行进度,半个小时啥运行信息都没有,体验不好。
- 数据为什么会出现这种长度片段不一的情形?
cellranger 系列软件使用说明
为了解决这个bug,cellranger-arc, cellranger, cellranger-atac这一系列的软件都用了一遍,因此简单记录下相关使用流程。
cellranger-arc
cellranger-arc 是10X开发的处理atac+rna的单细胞多组学数据的软件,先写cellranger-arc的原因是cellranger-arc的参考基因组可以用于cellranger, cellranger-atac,这样后面俩就不用再写怎样制备参考基因组了。
cellranger-arc mkref
这一步是制备参考基因组,即根据gtf(基因组注释文件)去制备参考基因组,告诉软件片段和基因的对应关系。基本命令如下
cellranger-arc mkref --config=/temp/swap/sunrui/genome/csp.config --nthreads=16
运行前需要有一个物种基因组的gtf注释(小鼠和人的10X提供基因组注释,其他的要自己拿gtf去做)。之后写一个config文件,告诉程序一些运行参数,config如下
{
organism: "csp"
genome: ["csp"]
input_fasta: ["/temp/swap/sunrui/genome_fa/csp.fasta"]
input_gtf:["csp.gtf"]
}
对gtf注释文件,有时候合作方会提供gff文件,这时候需要预先做一个格式转换,gff转gtf的工具,推荐使用agat。
cellranger-arc count
制备完参考基因组后,就可以进行count操作了,cellranger-arc count基本命令如下:
cellranger-arc count --id=CYBQ \
--reference=/temp/swap/sunrui/genome/mfu \
--libraries=CYBQ.csv \
--localcores=48 \
--localmem=48
其中id 是结果保存目录的名称,reference是上一步制备参考基因组所在文件夹,libraries是一个csv文件的路径,csv文件中记录了我们的.fq.gz测序数据的路径。最后两个参数是并行数目和分配内存,有资源就狠狠的加。
对于csv文件的写法,基本形式如下
fastqs,sample,library_type
/work/swap/sunrui/rna_seq/clean_rna/CY/fCY-BQ-G,fCYBQ,Gene Expression
/work/swap/sunrui/atac_seq/clean_data/CY/CY-BQ-A-1,CYBQ,Chromatin Accessibility
/work/swap/sunrui/atac_seq/clean_data/CY/CY-BQ-A-2,CYBQ,Chromatin Accessibility
/work/swap/sunrui/atac_seq/clean_data/CY/CY-BQ-A-3,CYBQ,Chromatin Accessibility
/work/swap/sunrui/atac_seq/clean_data/CY/CY-BQ-A-4,CYBQ,Chromatin Accessibility
第一列是.fq.gz测序路径文件夹的路径,第二列是这个路径下数据的名称,第三列是告诉程序这是ATAC还是RNA。
对于10X相关程序,其文件命名有特殊格式,记录如下:

可以看见文件命名都是按照这种规则进行的
sample_S1_L00?_??_001.fastq.gz
其中需要修改的有sample, 这里一个目录下的文件都要命名成一个sample。还有L00?,这个L代表lane的意思,我理解的就是样本是分了几条管道去做的,这个L00?告诉我们这组数据来自哪个管道,同样一个目录下的文件都要命名成一个管道。最后??,可选的是I1,R1,R2,R3。
以上是cellranger-arc 的使用说明
cellranger
cellranger是10X开发的针对单细胞RNA数据的分析流程,使用说明如下:
完成参考基因组制备后,运行下面指令进行count
cellranger count --id CYHM --fastqs CY-HM-G --transcriptome /temp/swap/sunrui/genome/csp
与cellranger-arc count不同的参数有, –fastqs,这里记录的是样本的.fq.gz路径,–transcriptome是参考基因组路径。
cellranger-atac
cellranger-atac是10X开发的针对单细胞ATAC数据的分析流程,使用说明如下:
cellranger-atac count --id=CYHM \
--reference=/temp/swap/sunrui/genome/mfu \
--fastqs=/work/swap/sunrui/atac_seq/clean_data/CY/CYHM \
--localcores=48 \
--localmem=64 \
这里的–reference是参考基因组路径。
fastp, agat使用说明
这两个软件在上面都有提到,这里不再仔细记录相关的指令,仅提供对应的github地址供后续查阅
fastp: https://github.com/OpenGene/fastp
agat: https://agat.readthedocs.io/en/latest/