本篇博客记录自己对24年Nature Methods上的文章 Toward universal cell embeddings: integrating single-cell RNA-seq datasets across species with SATURN 的解读。文章的通讯作者是 Jure Leskovec,图神经网络方向的大佬。解读这篇文章的目的是因为自己最近也在思考单细胞数据跨物种进化相关的问题,这篇文章对我想到的一些问题提供了一些很有价值的回答,因此记录下来。
概述
研究背景
- 跨物种的单细胞分析提供了进化分析的新的角度,现有方法缺乏跨物种单细胞数据整合方法
- 当前数据整合方法依赖一一对应的同源基因,但在多物种的情形下,会有大量非保守的基因,仅选择一一对应的同源基因会缺失大量信息。
主要结果
- 提出了SATURN的方法,该方法解决了跨物种的非同源基因数据整合问题,提供了单细胞分析与进化研究结合的新思路。
- SATURN给出了多组学研究的新思路,在单细胞分析中应用蛋白质语言模型进行建模。
- 引入宏基因的概念,并成功应用在多个数据集分析中。
模型
输入
- 多个物种的scRNA-seq数据,物种$i$数据记为$X^{s_{i}}$, 维度为$(C_{s_{i}},\vert \mathcal{G_{s_{i}}} \vert )$。$C_{s_i}$是物种的细胞数目,$\mathcal{G_{s_{i}}}$是物种的基因集合
- 细胞注释 $y^{s_{i}}$,维度为$C_{s_{i}}$
- 蛋白质嵌入 $P\in R^{\vert \mathcal{G}\vert \times p}$, 其中 $\mathcal{G} = \cup_{s_{i}} \mathcal{G_{s_{i}}}$, 记录了所有物种中各个基因对应蛋白质的向量表示。
输出
- 多物种细胞的统一嵌入 $z_{c}\in R^{k}$
- 基因-宏基因权重矩阵 $W \in R^{\vert \mathcal{G}\vert \times \vert \mathcal{M}\vert }$
宏基因(Macrogene)初始化
为了处理非同源基因,需要将不同的基因$\mathcal{G_{s}}$映射到公共的宏基因空间$\mathcal{M}$。宏基因中是通过蛋白质的信息定义的,而蛋白质的信息是由编码它的基因组序列信息决定的,也就是最基础的ATCG碱基序列,这是在所有物种中通用的。SATURN 通过一个非线性变换来实现,记$X_{c}^{s}$为物种$s$中的细胞$c$的基因表达数据,其对应的宏基因表达数据为
\[e_{c} = ReLU(LayerNorm(W_{s}^{T} \log(X_{c}^{s} + 1 )))\]这里$W_{s}^{T} \in R^{\vert \mathcal{M}\vert \times \vert \mathcal{G_{s}}\vert }$, 是一个物种特异的转换矩阵,也是后面网络学习的参数之一。
一个有生物解释性的宏基因的初始化方法是借助蛋白质嵌入信息实现。通过对蛋白质进行聚类,将其划分为M个具有一定生物意义(功能相似,序列相似……)的cluster,文章中通过KNN进行聚类。
之后将质心定义为宏基因,每个基因$g$到每个质心(宏基因)$m$的距离$d_{m,g}$反映了该基因与这个宏基因的关系,进一步定义转换矩阵$W \in R^{\vert \mathcal{G}\vert \times \vert \mathcal{M}\vert }$为:
\[W_{g,m} = 2\times(\log(\frac{1}{rank(d_{m,g})} + 1))^{2}\]前面提到的$W_{s} = W[\mathcal{G_s}, :] \in R^{\vert \mathcal{G_{s}}\vert \times \vert \mathcal{M}\vert }$
Autoencoder预训练
网络中用到一个基本的模块,记为$U$,定义如下
\[U(x) = Dropout(LN(ReLU(FN(x))))\]这里 FN 为全连接网络, LN为Layer Norm 。
encoder
encoder 部分, 将$X_{c}^{s}$ 映射到隐层表示 $z_{c} \in R^{k}$,网络结构为
\[X_{c}^{s} \in R^{\mathcal{G_{s}}} \rightarrow e_{c} \in R^{\mathcal{M}} \rightarrow U_{1}(U_{2}(e_{c}))=z_{c} \in R^{k}\]decoder
decoder 部分, 输出 $\mu_{c}, O_{c}, \theta \in R^{\vert \mathcal{S}\vert }$, 其中$\theta$是nn.Parameter() 定义的一个可微参数
\[\begin{aligned} \mu_{c} &= Softmax(W_{s}U_{\mu}U_{s}(z_{c}))*sum(X_{c}^{s}) \\ O_{c} &= FN(U_{s}(z_{c})) \end{aligned}\]损失函数
预训练部分损失函数包含两部分,第一部分为重构损失,刻画生成的数据与原始数据分布的差异。第二部分损失函数控制了$W$与蛋白质嵌入空间的一致性,因为初始$W$中的权重刻画了基因-宏基因在蛋白质空间中的一致性,希望训练后$W$仍旧保持这个特性。
重构损失是ZINB的似然估计,ZINB(zero inflated nonegative binomial)是一个常用的建模scRNA-seq数据分布的概率分布。zero flated 通常用来刻画以下情形:
- 若发生case 1: 计数为 0
- 若发生case 2: 计数过程服从某个概率分布
一般的负二项分布定义如下:
\[\begin{equation} P(X = k) = \binom{r-1}{k+r-1} p^{r}(1-p)^{k} \quad k=0,1,2,... \end{equation}\]ZINB定义如下: \(\begin{aligned} \hat{P}(X = 0) &= \pi + (1-\pi) P(x=0) \\ \hat{P}(X = k) &= (1-\pi) P(x=k) \end{aligned}\)
文章中的重构损失定义如下
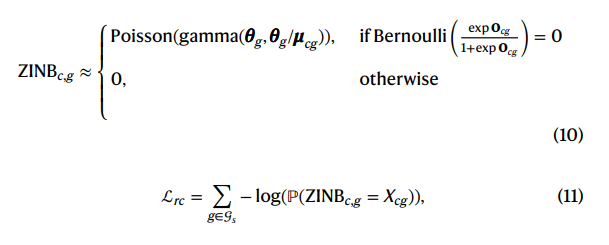
一致性损失定义如下

这里 $B = U(W) \in R^{\mathcal{G}\times p}$, $sim(. \; ; \;.)$是余弦距离。对$B,P$做相同的随机行重排,希望基因-宏基因矩阵$W$能够恢复蛋白质空间中的距离关系。
最终的损失如下
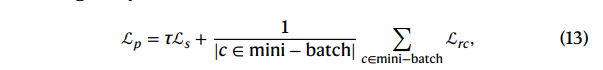
度量学习微调
使用度量学习的方法微调细胞嵌入$z_{c}$,此时仅调节encoder部分权重,decoder, $W$权重固定。度量学习的基本目标是控制$(anchor(z_{a}), positive(z_p), negative(z_n))$ 三元组之间的距离,使用的损失如下:
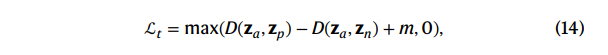
一个好的表示应当使得$z_a, z_p$之间距离小于$z_a,z_n$之间距离,因此在上面损失中,若$D(z_a,z_p) \geq D(z_a,z_n)$,loss会大于0,最小化loss使得$D(z_a,z_p) \leq D(z_a,z_n)$ 。
文章中关于$(z_a,z_p,z_n)$的选取如下:
- 选定物种特异细胞作为$z_a$
- $z_p$与$z_a$互为最近邻, 即在物种1中选择锚点细胞$x$,在其他物种中找到$x$的跨物种最近邻$y$,之后对$y$在物种1中选择最近邻$\hat{x}$,如果$x, \hat{x}$的细胞注释一致,则将$x,y$作为一对(anchor,positive)
- $z_n$选择为与(anchor,positive)细胞注释不同的细胞
Remark 1
度量学习中,必须要使用物种特异的细胞类型确定锚点吗?如何统一不同数据集细胞注释的标准?
实现细节
数据集预处理
使用公开数据集 Tabula Sapiens, Tabula Microcebus,Tabula Muris, 总共有335000个细胞
- 选择超过350个细胞的细胞类型
- 过滤表达基因数目低于500的细胞
- 过滤在少于1000个细胞中表达的基因
对frog(97000 cell) and zebrafish(63000 cell) embryogenesis 数据
- 过滤表达基因数目低于500的细胞
- 过滤在少于10个细胞中表达的基因
对所有数据进行HVG筛选,保留8000HVG,最终得到筛选后的count 数据。
蛋白质嵌入向量
使用ESM2模型生成表示,输入氨基酸序列,输出5120维向量嵌入。对编码多个蛋白质的基因,向量表示为各个蛋白质表示的均值。
Remark 2
怎样确定基因编码的蛋白质序列信息?直接通过外显子序列吗,这样会对基因组质量有很高的要求吧,要注释的足够准确。
超参数
宏基因组数目设置为2000,细胞嵌入和隐藏层的维度都是256
结果
创建多物种细胞图谱
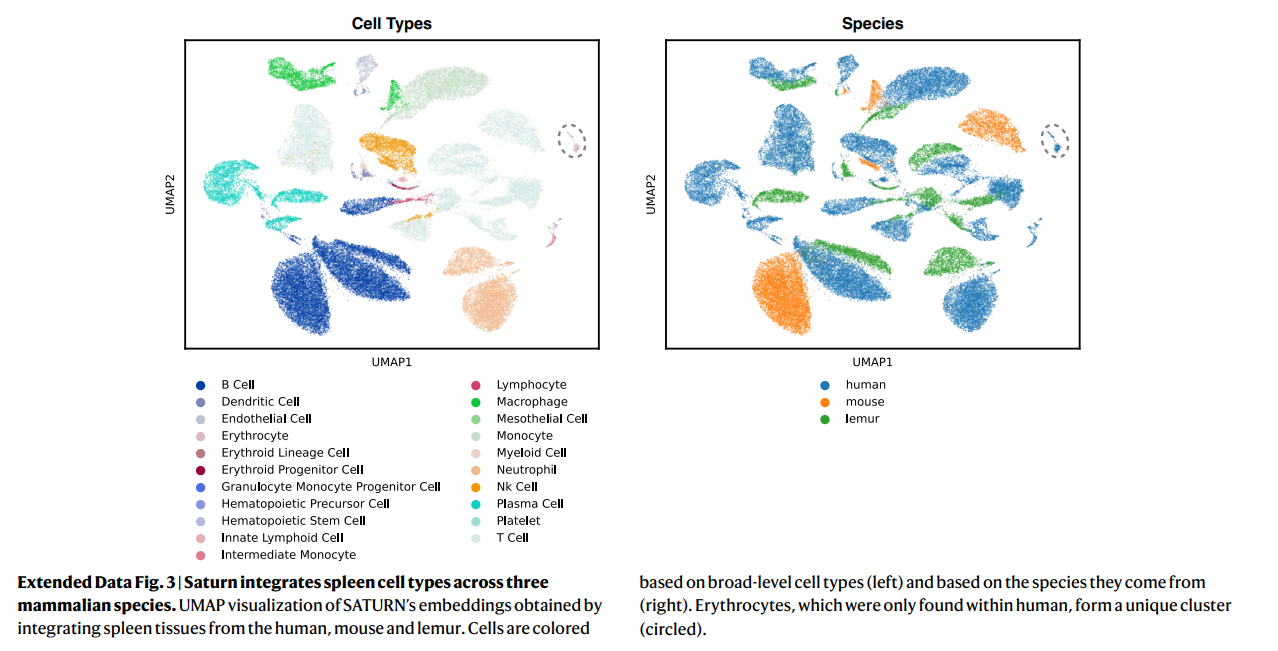
整合人类、小鼠、鼠狐猴数据,不同物种中相同的细胞类型能够得到很好的对齐,说明这些细胞具有进化保守性。
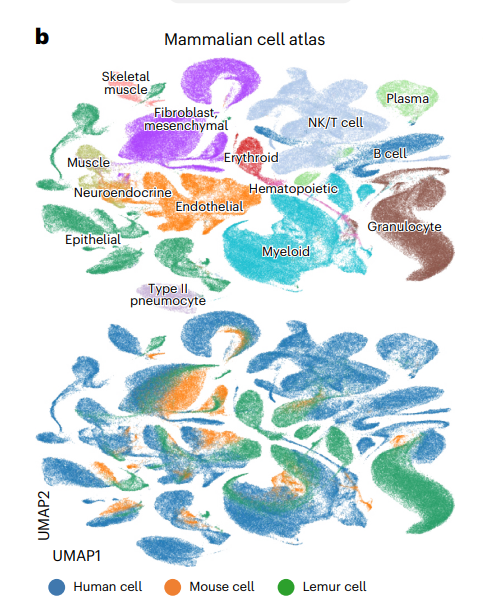
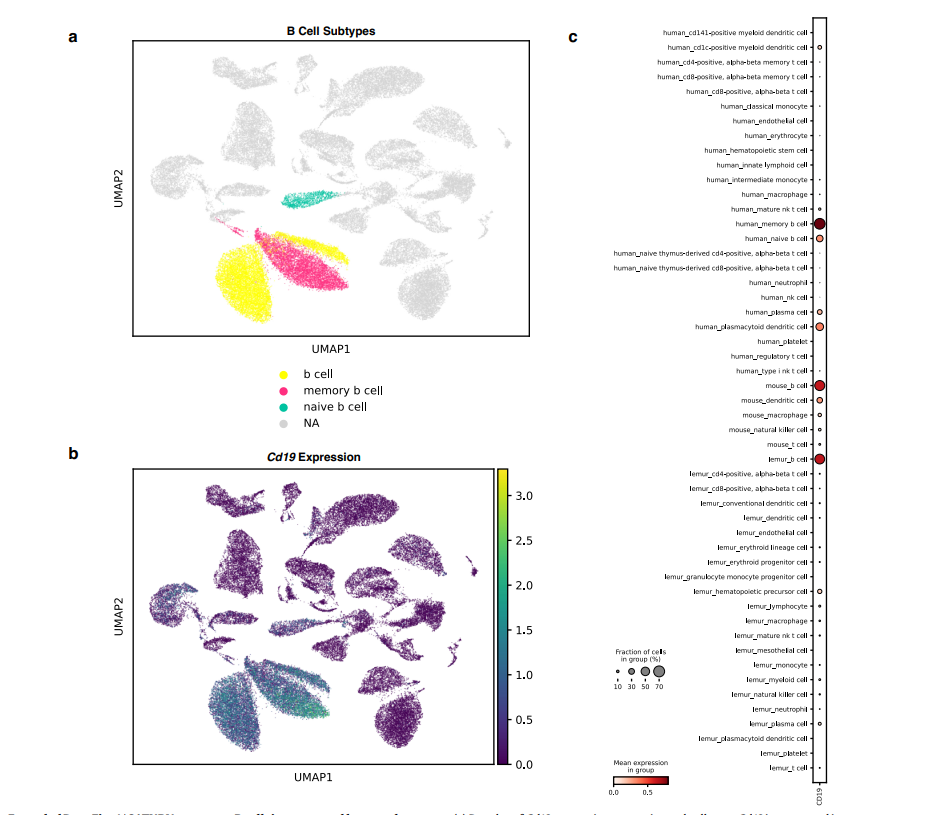
脾脏中,人类naive B细胞和memory B细胞被分隔开,但是naive B与鼠,鼠狐猴中B细胞对应很好
对这一点,可以通过基因Cd19的表达看到。在人类naive B细胞中低表达(cellMarker数据库中有收录,但是并未细分naive B 和 memory B,看起来是一个可信的结果)。
整合青蛙和斑马鱼的胚胎发育细胞
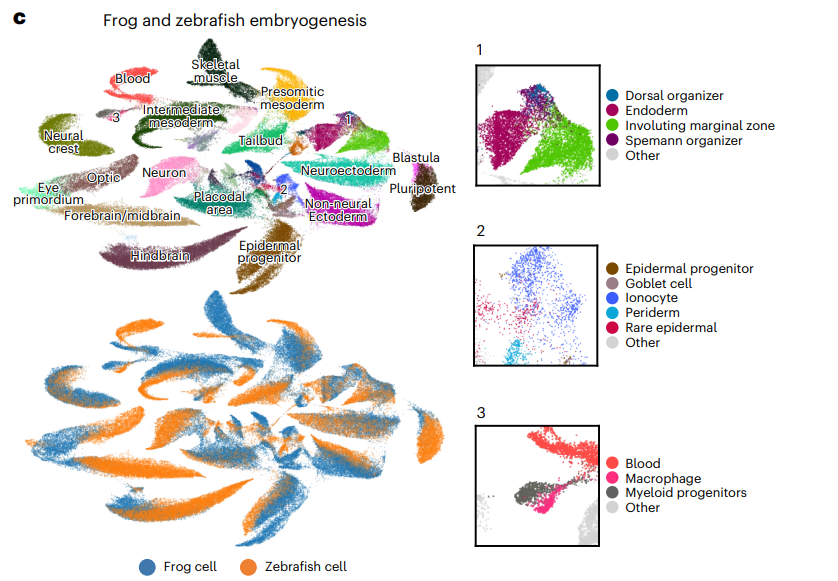
两个进化距离很远的物种能够得到很好的匹配,细胞注释是使用cluster中占多数的细胞注释。右侧小图展示了一些类型混杂的小的cluster,但可以发现这些混杂的细胞类型通常都有相似的功能。例如3中斑马鱼的巨噬细胞(macrophage)和青蛙的骨髓祖细胞(myeloid progenitors)被聚到一起,但两类细胞的后续分化过程中都涉及到Cybb,Cyba,Spib,Cepba这些保守的调控基因,对上述对齐给出了一个合理的解释。
在宏基因组层面分析差异表达
在宏基因表达数据上做scRNA-seq类似的差异表达分析,用宏基因中权重最高的基因来解释宏基因的功能。
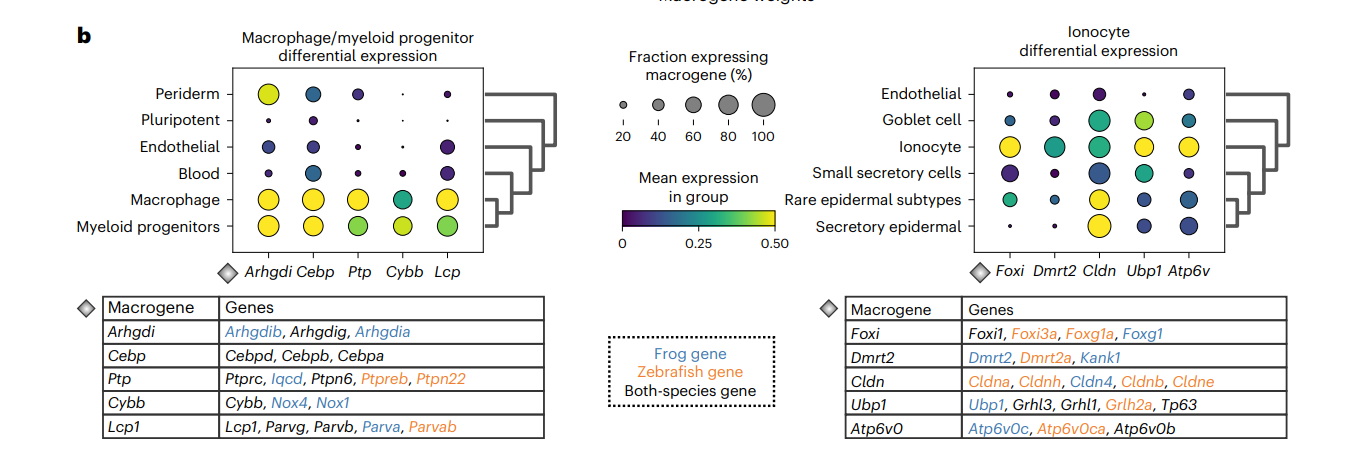
上图中的高表达宏基因,包含了相应的细胞特异基因,说明宏基因的生物学意义。例如左边的巨噬细胞/骨髓祖细胞中,相比其他各个细胞类型,宏基因都是高表达的。
宏基因也具有物种特异性,例如下图中的宏基因都是在物种内特异表达的

宏基因保持了序列同源
对每个宏基因,统计各个物种的基因在这个宏基因中的权重,每个物种选取Top-k个基因,统计Top-k 基因中包含一对同源基因的宏基因比例,在青蛙和斑马鱼中结果如下
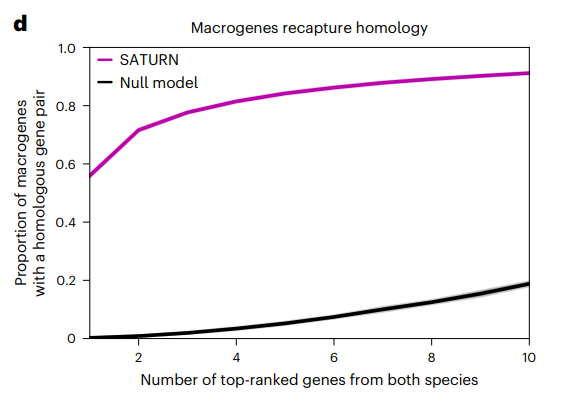
其中的Null model 是每个物种的基因随机排序后,Top-k基因中包含同源基因的宏基因比例。可以看到宏基因很好的保持了序列的同源性。
在小鼠和人的基因组中进行Gene Ontology enrichment analysis,可以验证宏基因很好的保持了基因功能的相似性
SATURN 方法评估
通过标签迁移来评估嵌入的有效性
使用SATURN整合数据,之后在物种保守的细胞类型上,在一个物种上训练一个logistic regression分类器,在另一个物种上做预测,统计能够分类的准确性。比起其他方法,SATURN迁移的准确性更高。
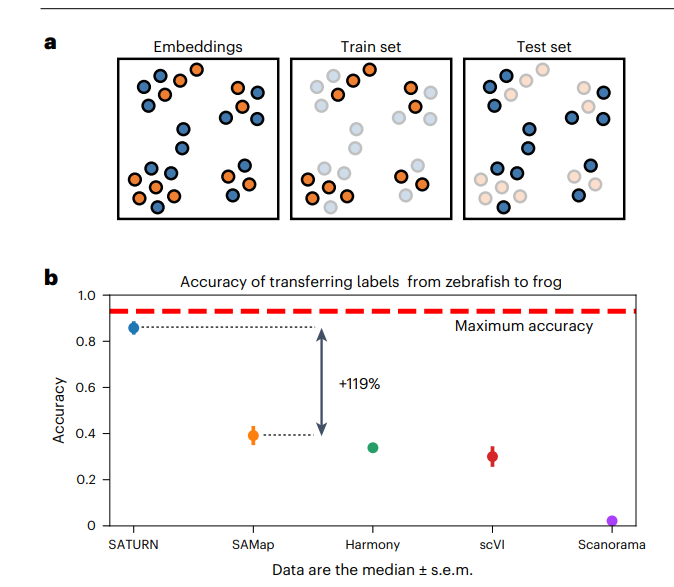
Remark 3
其中SAMap是考虑了非同源基因的方法,Harmony,scVI,Scanorama未考虑非同源基因。Harmony, Scanorama这两个方法好像都是用了基于锚点的整合策略,这么大的差异挺奇怪的。
对比起来看,考虑非同源基因是一个挺有效的策略。下图是各个方法整合效果的对比

对Harmony,scVI,Scanorama对齐表现这么差的情况,我保持怀疑,个人在猪尾鼠和小鼠上没看到这么大差异,可能的原因是青蛙和斑马鱼进化差异更大?同源基因更少?
SAMap对齐效果看起来更好,但是在细胞类型的保守性上表现较差。
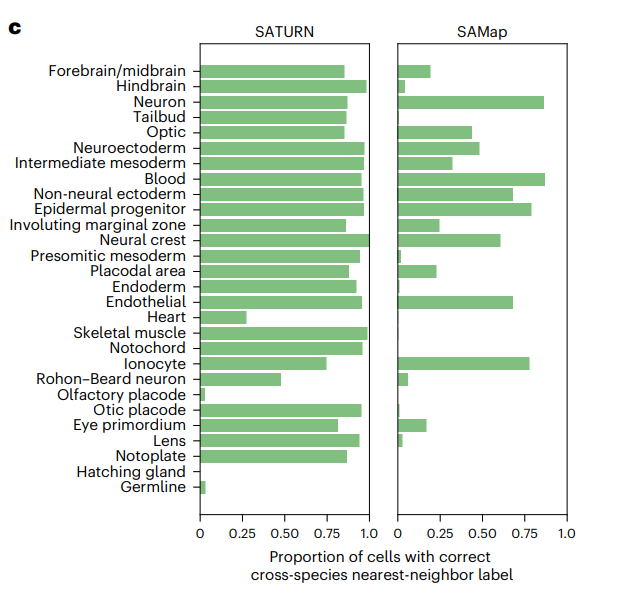
SATURN 更好保留最近邻细胞信息
下图展示了在物种保守的细胞类型中,对每一个细胞类型中的每一个细胞,查找其跨物种最近邻细胞,统计跨物种最近邻细胞与该细胞类型一致的比例。总体来看SATURN表现不错,对于数目较少的细胞类型表现较差。
SATURN 整合AH altas中的五个物种
验证方法在多物种处理上的有效性,从结果上看保守性做的不错
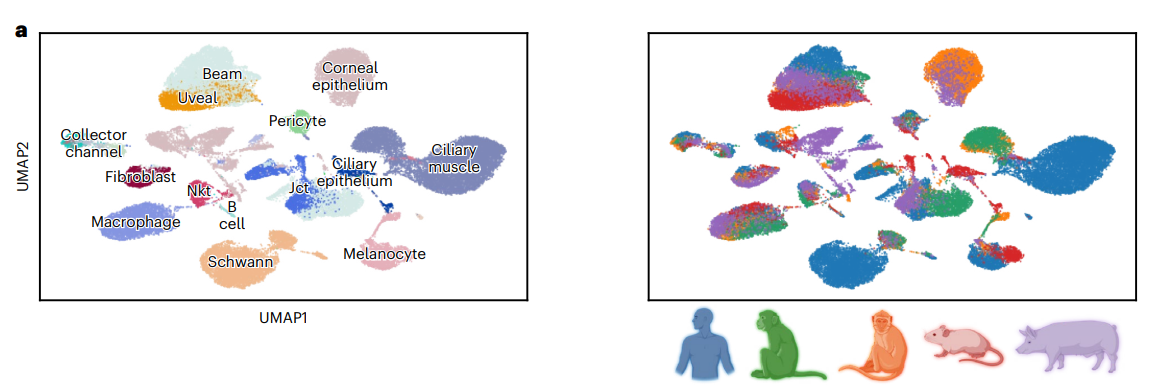
对原有注释中保守性较差的21个细胞类型进行重聚类
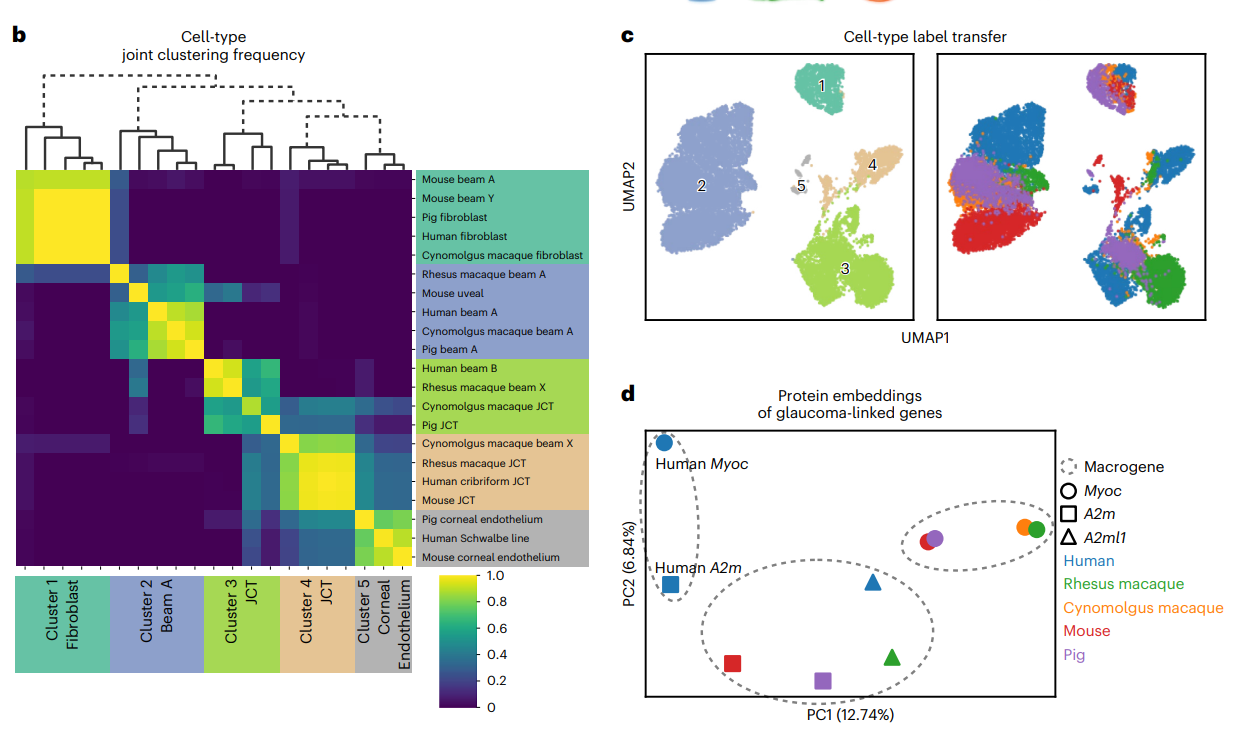
SATURN 预测同源基因的不同功能
上图中子图d 展示了物种中青光眼相关基因,人类的Myoc基因和其余四个物种的Myoc基因相隔很远,同时,人类的Myoc基因所在的宏基因中的top 1基因为A2m,可以看到和其他四个物种的A2m基因相隔很远。而Myoc和A2m是序列差异很大的。
因此,蛋白质序列嵌入可以帮助分析相同基因在不同物种中的功能差异,以及相同物种内序列差异较大的基因在功能上的潜在相似性。
总结
亮点
- 蛋白质语言模型应用到基因组学分析中,给出了多组学建模的新思路
- 实现跨物种非同源基因的整合,充分利用了信息
- 宏基因的引入,增强模型的解释性
- 给出了单细胞分析和进化生物学研究结合的策略
局限
- 需要参考蛋白质组,对非模式物种来讲比较难获取
- 单基因编码多种蛋白质的情形,使用简单的均值作为基因表示
- 模型需要细胞注释信息,细胞注释本身存在误差。
对我个人来讲,两个我关注的问题都得到了一定程度的解决,给后续的研究提供了思路,一是单细胞分析和进化研究的结合,怎么分析,可以挖掘哪些信号,文章的分析可以借鉴。二是非同源基因的整合,文章用蛋白质语言模型的方法建立了非同源基因之间的联系,同时也给出了大规模预训练模型在下游分析中应用的范例。非常漂亮的工作!