最近在做一份大模型的实习,了解了LLM相关知识后回头看下生信方向,尤其是单细胞方向大模型的进展。这次的论文是scGPT: toward building a foundation model for single-cell multi-omics using generative AI。论文是2024年1月份被Nature Methods接收的,但是2023年的时候就挂在biorxiv上了,取的名字scGPT也算是小蹭了一波OpenAI的热点吧。
概述
研究背景
生成预训练模型在自然语言(GPT)以及计算机视觉(DALL-E)都取得了巨大成功,尝试将类似的方法迁移到单细胞研究中,建立foundation model,尝试统一单细胞研究领域task-specific的研究范式。
主要结果
- 构建了scGPT模型(参数量38M),验证了foudation model在单细胞领域的可行性
- 构建了一个大规模预训练数据集以及多样的下游微调数据集。
- scGPT成功应用到多个下游任务中并取得不错的性能。
数据
scGPT 整合了一个大规模的预训练单细胞数据集以及多样的下游微调数据集资源,这也是文章的一个重要贡献。下面对用到的数据集进行一个总结。
| 数据集 | 数据规模 | 数据类型 | 数据任务 | 数据地址 |
|---|---|---|---|---|
| CELLxGENE | 33M | scRNA-seq, snRNA-seq | 预训练数据 | https://cellxgene.cziscience.com/ |
| Multiple sclerosis | 22000 | 细胞类型注释 | https://www.ebi.ac.uk/gxa/sc/experiments/E-HCAD-35 | |
| Myeloid | 13000 | 细胞类型注释 | GSE154763 | |
| Human pancreas | 15000 | scRNA-seq | 细胞类型注释 | Transformer for one stop interpretable cell type annotation |
| PBMC 10K | 12000 | scRNA-seq | scRNA-seq整合 | A Python library for probabilistic analysis of single-cell omics data. |
| Immune human | 33506 | scRNA-seq | GRN推断 | Benchmarking atlas-level data integration in single-cell genomics |
| Perirhinal cortex | 17500 | scRNA-seq | scRNA-seq整合 | Transcriptomic diversity of cell types across the adult human brain. |
| COVID-19 | 274346 | scRNA-seq | scRNA-seq整合 | Mapping single-cell data to reference atlases by transfer learning |
| Adamson | perturb-seq | 基因扰动预测 | A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response | |
| Norman | perturb-seq | 基因扰动预测 | Exploring genetic interaction manifolds constructed from rich single-cell phenotypes | |
| Replogle | 171542 | pertub-seq | 基因扰动预测 | Mapping information-rich genotype–phenotype landscapes with genome-scale Perturb-seq |
| 10x Multiome PBMC | 9361 | scRNA-seq,scATAC-seq | 单细胞多组学整合 | https://support.10xgenomics.com/single-cell-multiome-atac-gex/datasets/1.0.0/pbmc_granulocyte_sorted_10k |
| BMMC | 90261 | scRNA-seq, CITE-seq | 单细胞多组学整合 | A sandbox for prediction and integration of DNA, RNA, and proteins in single cells |
| ASAP PBMC | 17000 | gex, chromatin accesibility, proten abundance | 单细胞多组学整合 | Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells |
| Lung-Kim | 32493 | https://www.weizmann.ac.il/sites/3CA/lung |
模型
模型结构
scGPT的结构和常见的LLM类似,包含12个transformer block, $d_{model} = 512$,模型参数约38M。从scGPT代码中看LN用的好像是DSBN,DomainSpecificBatchNorm.
不同于GPT的decoder only 架构,scGPT还是经典的Transformer encoder-decoder架构,更类似于masked language model,下面的源码也可以看到明显的encoder,decoder架构。
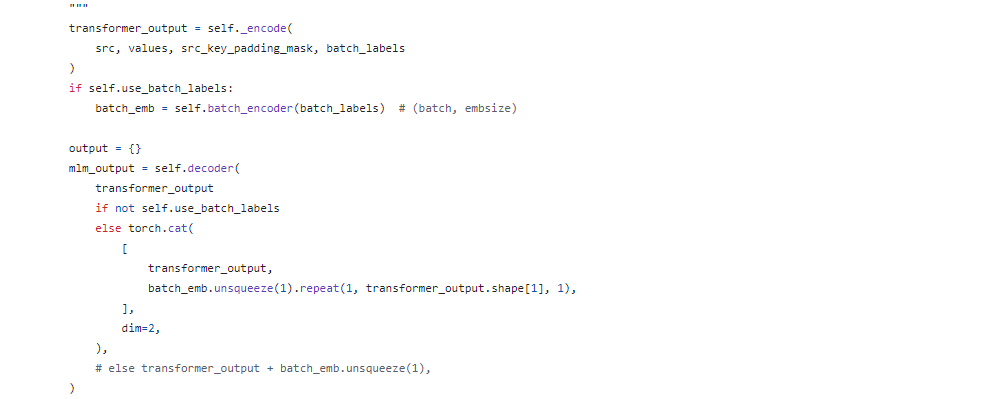
Remark1:
从源码中看,scGPT是没有使用位置编码的,这个我认为是合理的。NLP中使用位置编码是因为一个单词可以出现在一句话的不同位置,加入位置编码来区分不同位置的不同单词。但在单细胞任务中,不会出现同一个基因在一个细胞中的不同测量,因此没有必要使用位置编码。
Tokenization:
使用的token为gene和condition。gene token为每个基因分配一个整数索引$id_{g}$, condition token为细胞的每个condition(例如扰动某个基因)分配一个整数索引$id_{c}$。每个细胞tokenization后会得到三组向量:
\[\begin{equation} \begin{aligned} gene_{id} &= \{id_{g_{1}}, id_{g_{2}},..., id_{g_{M}}\} \\ gene_{exp} &= \{bin(g_{1}), bin(g_{2}),..., bin(g_{M})\} \\ condition_{id} &= \{id_{c_{1}}, id_{c_{2}},...id_{c_{M}}\} \end{aligned} \end{equation}\]其中$gene_{exp}$是作者考虑不同数据集中细胞的基因表达存在技术差异,很难通过TPM标准化或者$\log(x+1)$变换去消除这种差异,因此对每个细胞的非零基因表达进行分箱(bin)操作。分箱操作是对单个cell进行的,不同的cell的划分$[b_{k},b_{k+1}]$不同。
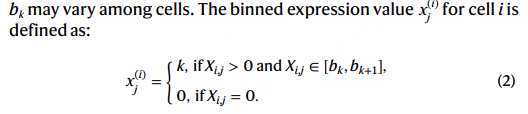
特殊的token有 $
Embedding
$gene_{id}, condition_{id}$的嵌入是通过nn.Embedding()定义的,$gene_{exp}$通过全连接网络得到一个相同维度的嵌入,最终单个细胞的表示为一个(M,D)大小的张量,M为token数目(gene数目),D为单个token的嵌入维度: \(h = embed_{g}(gene_{id}) + MLP(gene_{exp}) + embed_{c}(condition_{id})\)
cell representation
如前所述,细胞的表示为encoder的最后一层的表示$h^{n} \in R^{M\times D}$中
batch 和 modality信息的处理
对于batch或者modality信息,scGPT没有在预训练过程中加入相关的信息,而是在下游微调任务中加入对应的batch token或者modality token,直接将scGPT的输出拼接上对应的batch embedding或者modelity embedding进行后续微调
-
scRNA-seq scATAC-seq 整合
细胞c中的基因g表示为 \(h_{c,g} = concat(h_{c,g},emb_{b}(batch_{c}) + emb_{m}(modality_{g}))\)
-
scRNA-seq 整合
细胞c中表示为 \(h_{c} = concat(h_{c},emb_{b}(batch_{c}))\)
预训练任务
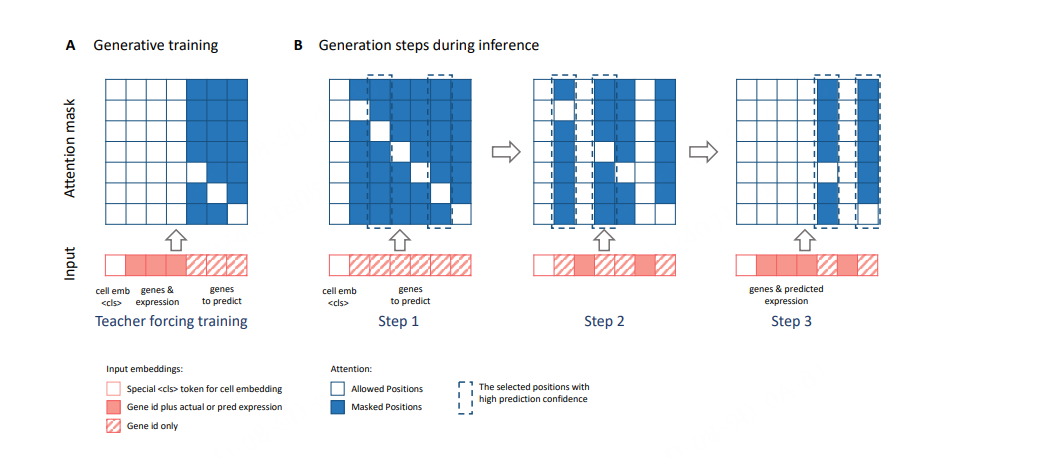
对于单细胞任务,关注的一个主要问题是,已知细胞状态或者部分基因表达的情况下,推断全基因组的表达量。
不同于NLP中next token prediction的预训练任务,细胞的基因表达量是没有顺序关系的,因此不能直接套用该任务。作者使用了Attention Score引入顺序关系。
具体一些,生成过程中,遮挡住的基因集合记为$G_{1}$,无遮挡的基因集合记为$G_{2}$,此时计算masked attention,attention矩阵中包含$G_{2}$中基因的两两注意力(三角矩阵)和$G_{1}$中基因自身的注意力(对角矩阵),之后根据$G_{1}$中基因的attention score选取top K基因,根据transformer decoder部分的输出预测这Top K基因的表达量。之后将这Top K基因从$G_{1}$中移除,加入$G_{2}$,重复上述操作直到全部基因预测完。(以上是我自己推测的生成过程,总之要引入顺序关系去做掩码预测,至于是用attentiion score排序还是再加几层网络预测一个置信度去选择,应该都合理)
最终的目标函数如下:
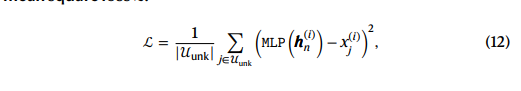
unk 代表未知的基因集合(待预测)。
下游微调目标函数
- 基因表达预测
和预训练目标基本一致,没啥好说的。
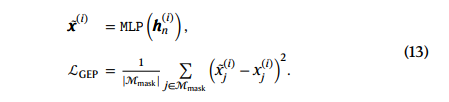
- 基于细胞嵌入的基因表达预测
和任务1不同的是,仅有细胞的嵌入向量$h_{c}$.
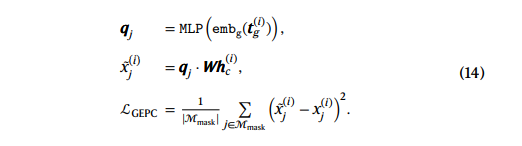
-
Elastic cell similarity
输入是两个细胞的嵌入向量,使得相似度超过阈值$\beta$的细胞更相似
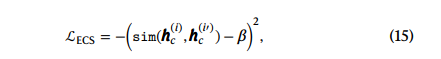
-
Domain adaptation via reverse back propagation
为了消除细胞表示中的batch effect,基于细胞表示,通过MLP分类器预测batch
-
细胞分类
根据细胞的表示做分类
下游微调任务
- 细胞类型预测
- 扰动预测: 基于control细胞预测扰动细胞的基因表达
- 多个scRNA-seq数据整合:使用Domain adaptation via reverse back propagation微调模型。
- scMultiomic数据整合:预训练时仅有gene token,加入多组学后扩增相应的token,之后继续预训练得到多组学下的细胞嵌入。
- 基因调控网络推断: 基于基因嵌入构建相似性KNN网络,之后leiden聚类提取gene programs。对于扰动数据的基因表达建模使用attention score去推断网络。
下游微调任务Benckmark
略。
结果
感觉没有特别多要说的,数据集都是公开数据集的原因,没有特别多新的生物上的结果。方法上的话就是和其他方法的对比,简单放点图和表格。
- 细胞类型注释
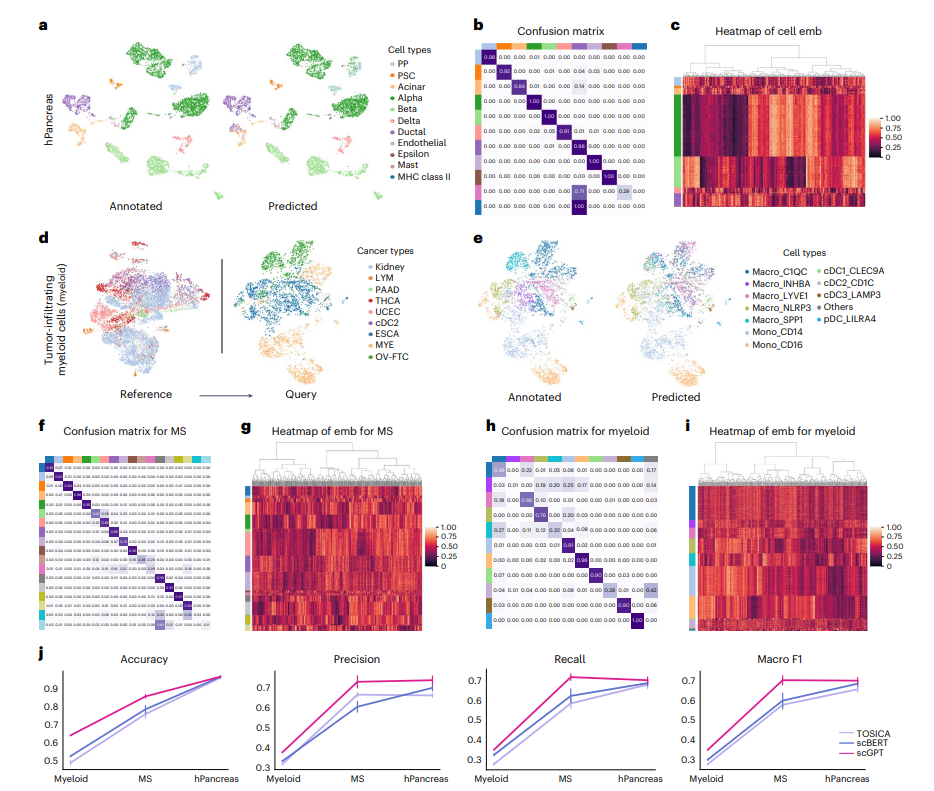
看图j,效果较其他方法好些
- 扰动预测
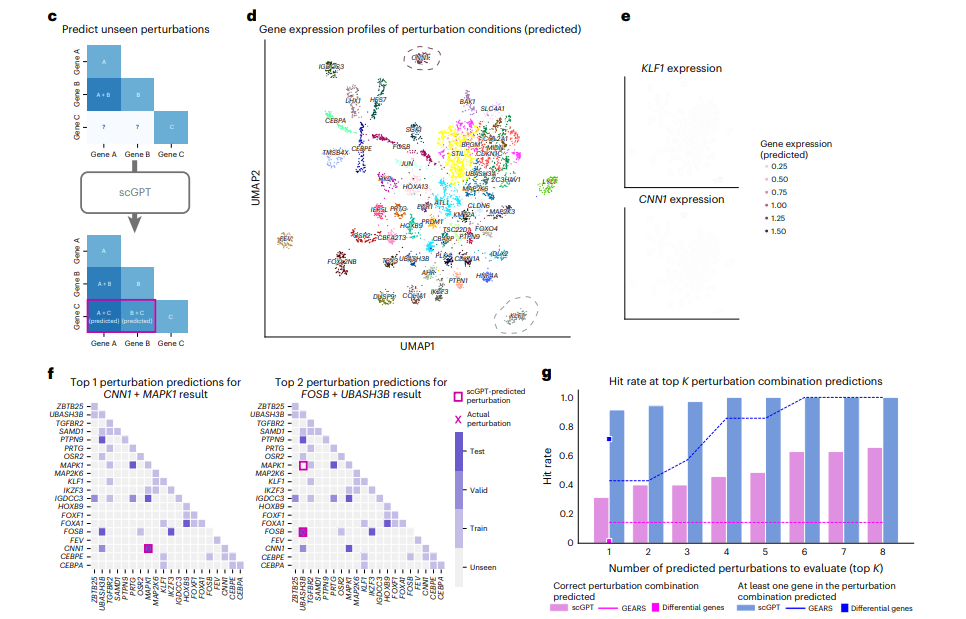
结果上优于GEARS
- 数据整合
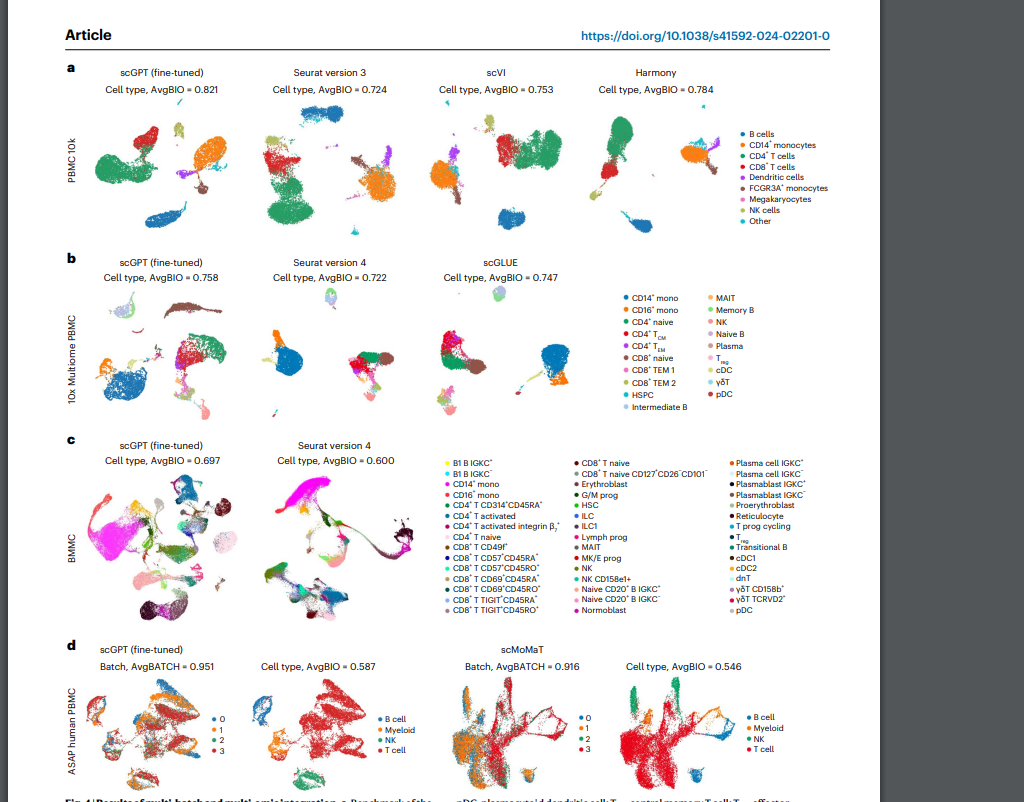
对齐效果由于其他方法。
总结
个人不是特别看好scGPT这个工作。从创新性来看,类似的单细胞大模型scBERT的发表在它之前,所谓的gpt看下来也并没有达到当下大热的chatgpt相当的level,至少在我看来它更像一个BERT类型的工作。同时38M的参数量也远未达到当下LLM的力大砖飞的地步。
从应用角度来看,在一些量化指标上看scGPT确实优于其他方法,但是从单细胞分析本身的应用来讲,这种量化指标的好不一定真的在应用层面上也更优。生信分析本质上是要讲好一个科学发现的故事,生物合作者更关注的是能否找到信号以及这些信号能否合理的有逻辑的讲好一个故事,至于方法本身的效果,在我看来0.80的精度还是0.70的精度没有什么本质上的差异。不是说提高算法精确性不重要,而是说准确性在生信分析上不是最核心的点。准确性很显然是需要一个benchmark来评估的,但是生信分析是在未知数据集上去分析,这种“已知-未知”的gap的存在,让人很难说benchmark上好的工作就更好。
从整个建模角度来看,我不太确信在单细胞这个任务上,transformer架构会显著优于其他的模型,比如说VAE。transformer架构是在大规模预训练之后才开始领跑NLP的,其核心优点是适用于大量的无标注语料的学习。而单细胞本身的复杂性和人类语言的复杂性相比,我个人是倾向于单细胞本身没有那么复杂,大量无标注语料预训练可能意义没有语言那么大,原因是单细胞技术本身的测量精度,注定了我们的观测时伴随着技术噪音的,分析时更多的会在cluster这个粒度下去看,这就大大减少了单细胞的复杂性。我觉得欠缺一个消融实验,比如说用一个大一点VAE在这种数据上训一遍看看差距。之前解读的SATURN就是用一个VAE框架做到的。
最后就是个人对单细胞领域大模型的一点看法,从我目前了解的一些工作,scBERT、scGPT、GeneFormer、Cell2Sentece等等,我并没有感觉到和NLP中LLM带来的同等级别的冲击。NLP中所有任务都被大一统成文本生成,在单细胞中这样的大一统任务看起来遥遥无期。总之,我不认为目前的这些单细胞大模型具备颠覆领域的能力,和之前的算法工作没有什么本质的区别。
当然,scGPT的工程量确实很大,整理的数据集也足够丰富,我个人还是很尊重这个工作本身的。前面提到的不看好仅是个人对单细胞大模型发展的一家之言,也许后面就被打脸了呢。