回声定位物种单细胞多组学数据分析工作总结
孙睿 2024.07.11
工作概况
这一部分阐述研究的主要问题和当前取得的一些结果。
研究问题
研究的问题是从分子层面解析回声定位物种的形成,是基于单细胞数据解读物种进化的研究工作。使用的数据是5个物种(长翼蝠,菊头蝠,犬蝠,小鼠,猪尾鼠)的各自的八个脑区(小脑半球,垂体,海马,脑干,皮层,丘脑,嗅球,小脑蚓部),总计40个样本的10X 多组学单细胞数据。其中小鼠和犬蝠是非回声定位物种,剩下三个物种是回声定位物种。
研究思路
- 掌握数据情况,对数据进行精确注释。
- 定位与回声定位性状最相关的脑区。
- 定位该脑区中与回声定位性状最相关的细胞类型。
- 对该类细胞类型进行跨物种比较,差异基因分析以及调控网络差异分析。
当前进展
- 完成40个样本的数据细胞类型的注释。
- 定位海马为潜在的回声定位性状相关脑区。(细胞比例差异)
- 完成各个脑区的调控网络推断工作。
主要难点
截止到目前,分析中存在主要的难点总结如下:
- 非模式物种的单细胞原始测序数据处理
- gtf文件修改
- 测序数据bug
- scRNA-seq 数据分析
- 跨物种数据整合
- 细胞类型注释
- scATAC-seq 数据处理
- scATAC-seq数据处理基本原理
- 调控网络推断流程
- Atlas 量级数据的分析
- 流程化处理
- 研究目标合理的“捉大放小”
-
数据备份
- 单细胞数据与性状结合的差异分析。
经验交流
下面针对前面提到的难点,记录自己的解决方案及相关的生物背景知识。
非模式物种,10X多组学数据基本处理
这一部分的难点在于,非模式物种的基因组通常会存在一些格式上的问题,这些格式问题会导致10X的数据处理流程出现问题。解决这些问题需要我们对10X数据的测序原理和相关的基因组文件格式有大概的了解,下面将描述我遇到的问题、基本的背景知识以及最后的解决策略。
基因组注释问题
遇到的问题是,对于非模式物种,10X cellranger-arc需要根据gtf格式的基因组注释文件来制备参考基因组,有了参考基因组才可以进行后续的count操作。而合作方仅提供gff格式文件,需要自行进行文件格式转换。
-
首先说明一下为什么需要基因组注释文件。
单细胞测序技术,实际上是测量了大量的序列片段。每个片段在测序时会附加一个barcode序列,barcode告诉我们这个片段来自于哪个细胞,而这个片段对应的分子信息则需要我们通过序列比对得到。
对于scRNA-seq数据,如果预先知道每个基因对应的mRNA序列,那么通过序列比对可以确定该片段对应哪个基因。对于scATAC-seq数据,如果预先知道整个基因组的序列信息,那么通过序列比对可以确定该片段是从基因组上哪个部分剪切下来的,从而确定peak。
那么进行序列比对需要什么信息呢?一是基因组序列,告诉我们每个位置的碱基信息。二是基因组注释,告诉我们基因组序列上这个区间是什么东西。有了这两个信息,我们就可以知道基因组上任意一个片段的碱基序列,从而可以和测量得到的序列进行比对。
-
接下来说明这些文件大概是什么样子。
基因组序列数据,通常保存在.fasta文件中,对每条染色体记录其全部的碱基序列。
基因组注释数据,通常保存在gff文或者gtf文件中。这两个文件是非常相似的,可以相互转化。这个文件提供的最主要信息是,具体格式请自行查阅资料。
|染色体|起始位点|终止位点|注释|对应基因| |—|—|—|—|—|
-
接下来说明我的解决措施。
gff转gtf的软件有挺多的,开始我使用的是cufflink里的gff2gtf的功能,但是转换后并不能用。所以后面我让一个代码大模型写了一段python脚本来转换,这个东西理论上并不难做,就是把一些列的信息进行调整即可。用这个方法我解决了猪尾鼠的gtf文件。
对于剩下的三种蝙蝠,这个脚本出了一些Bug,转换出的gtf文件出了一些顺序问题。具体一点就是,gtf文件并没有保留基因组顺序结构。出现了一些如下的bad case:
chr1, 10000, 11000 chr2, 200,2200 chr1, 100, 1100可以看到gtf文件中,染色体1上的注释还没有完成的时候,混入了一些其他染色体的注释,同时染色体1上的注释,在靠后的位置(10000-11000)的注释写在了靠前的位置(100-1100)的前面。一个合理的注释应该是这样的:
chr1, 100, 1100 chr1, 10000, 11000 chr2, 200,2200对这个情况,讲道理可以自己修改算法解决的,但我选择试一试比较新的转换工具,看看能否解决。比较幸运可以使用AGAT这个工具解决。
-
犬蝠的bug
这个问题的解决参见:https://moonbyul-99.github.io/2024/01/28/cellranger-error/
所以小结一下这个问题,完全是一个工程上的障碍,结合cellranger-arc的报错信息结合测序基本原理一点点修正就可以解决,但是如果没有解决过,还是会有点棘手,因此记录一下这个问题。
fastq数据问题
这个问题的解决参见我的博客内容:https://moonbyul-99.github.io/2024/01/21/cellranger/
scRNA-seq数据分析及整合
这部分工作主要有四部分:
- scRNA-seq 基本处理流程,见 https://moonbyul-99.github.io/2023/10/20/scRNAseq/
- 数据整合
- 细胞类型注释
- 差异分析
这里主要介绍2,3两点。这两项工作花了很长的时间,市面上常见的方法基本都试了一遍,是一段非常曲折的工作经历。
数据整合
做数据整合的原因是,需要给出40个样本的全局的展现,基于这个结果进行后续的分析工作。将40个样本的scRNA-seq数据,保留大约10k个同源基因后做降维可视化,得到结果如下:
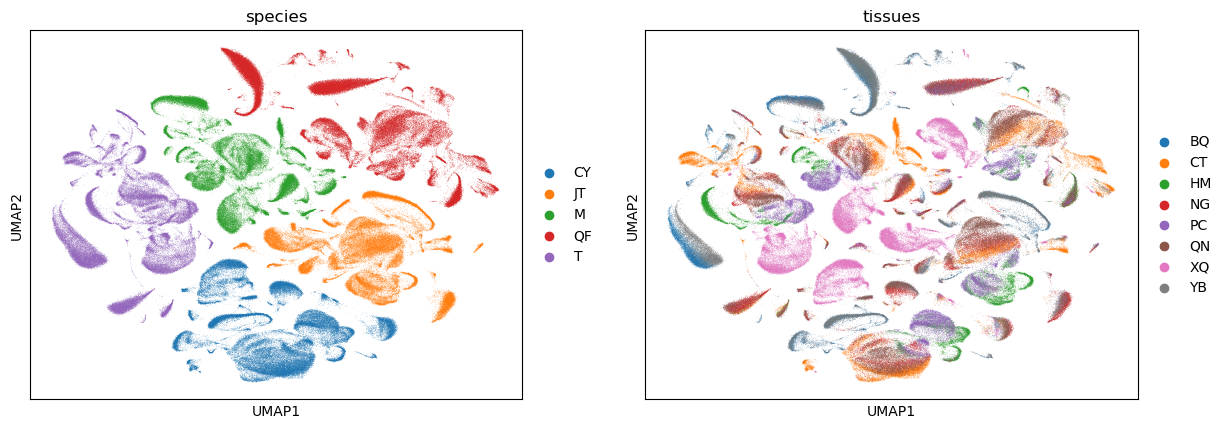
直接降维可视化结果,speices: CY,长翼蝠,JT,菊头蝠,M,小鼠,QF,犬蝠,T,猪尾鼠。tissues: BQ,小脑半球,CT,垂体,HM,海马,NG, 脑干,PC,皮层,QN,丘脑,XQ,嗅球,YB,小脑蚓部
可以看到不同物种的数据是完全没有重合的,在同一个物种内部,各个脑区也是界限相对清晰的。这个结果是不太合适的,因为我们的假设是一些细胞类型应当呈现跨物种的一致性(保守性)。而基于现在的这个UMAP结果,做细胞聚类每个cluster大概率都是物种特异的。因此需要对数据做整合。
数据整合需要明确以下几点:
- 整合什么数据
- 整合到什么程度(去除哪部分批次效应)
- 使用什么整合方法
对于整合的数据,这里选择对scRNA-seq数据进行整合。没有做多组学数据整合的原因是,市面上容易跑通的多组学数据整合的工作并不是特别多,我印象中发表在大子刊上、能够处理多样本多组学的整合方法可能就是totalvi, glue。
权衡下来决定不要投入大量时间在一个收益未知的方法上,scRNA-seq数据的整合方法一天就能尝试好多中,而多组学整合我可能要干半个月并且结果位置。丢弃atac-seq数据固然损失了部分信息,但能更快的推进工作。
确定做scRNA-seq数据整合后,我选择从对scanpy生态友好的方法中进行选择,尝试过的方法包括mofa+, harmony, scanorama, mnn, bbknn, scvi,meta-neighbor。具体的使用体验以后会讲。最后用的是harmony。
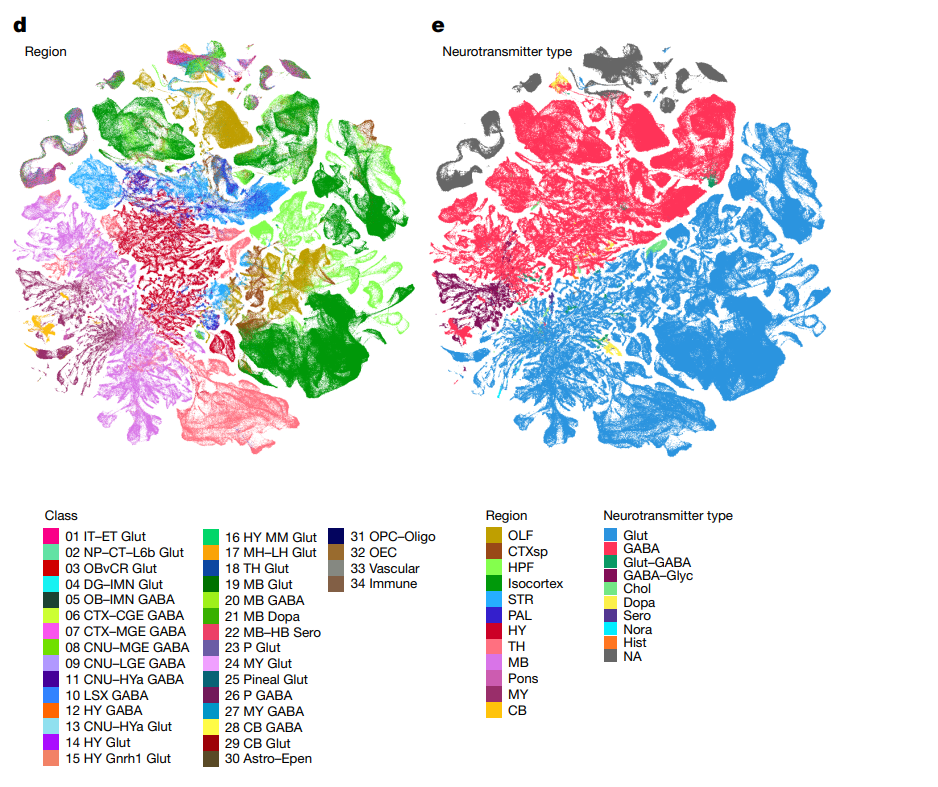
最关键的一点是要确定整合到什么程度,这个是要和生物学先验结合的。具体来说就是,是要让五个物种对齐(聚类后每一个cluster中都包含5个物种)还是让八个脑区对齐(聚类后每个cluster中都包含8个脑区)还是这40个样本对齐(每个cluster中包含这40个样本)。这个先验对于评估整合效果的好坏是至关重要的,前期因为缺乏先验,做了大量的失败的尝试。获取这类先验的方法是看文献,看下权威机构的类似工作是怎么整合的。后期我是参考的Allen Institute for Brain Science 在2023年发表的工作1。
这个工作做的是小鼠全脑的单细胞图谱,我们关注一下主图的结果d,可以看到UMAP展示是呈现脑区特异的。这提示我们整合对齐物种、保留脑区差异是一个合理的做法。
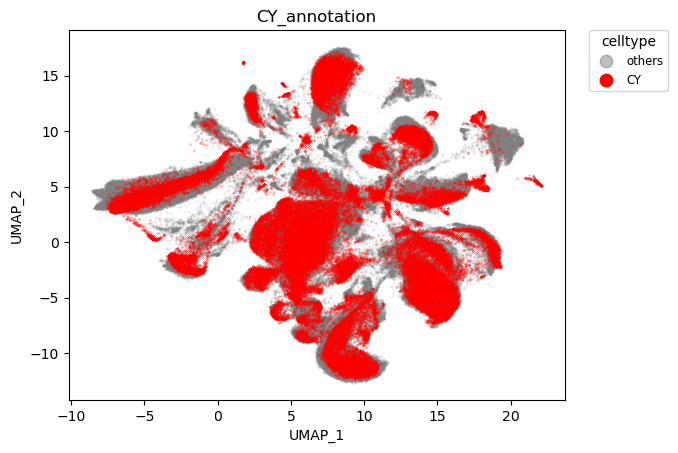
至此,数据整合中的几点都已明确,40个样本总共提供了454K 细胞,通过QC后保留339K细胞,使用harmony整合结果如下

harmony整合结果
可以看到,物种实现了对齐,而脑区则依旧保持特异性。
物种对齐效果:
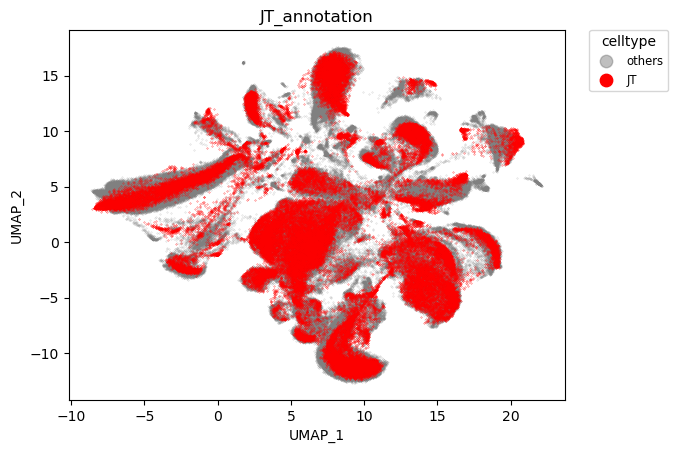
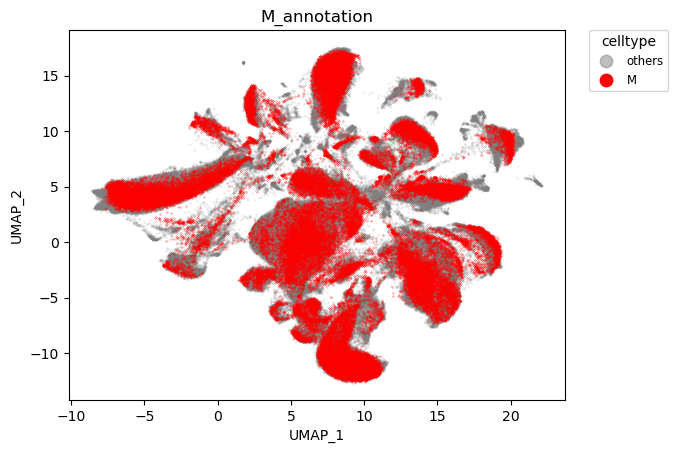
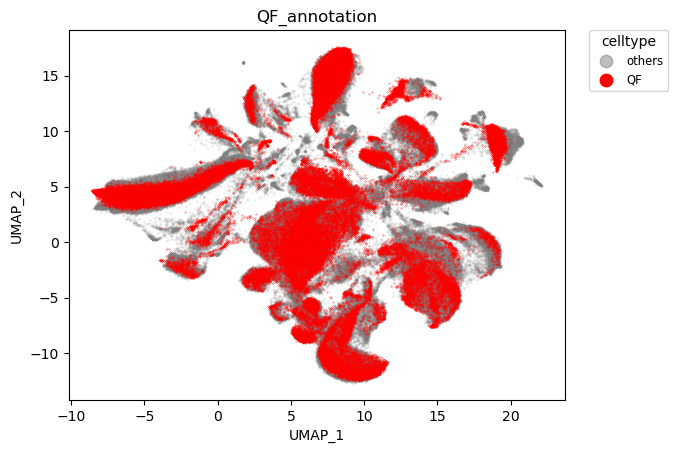
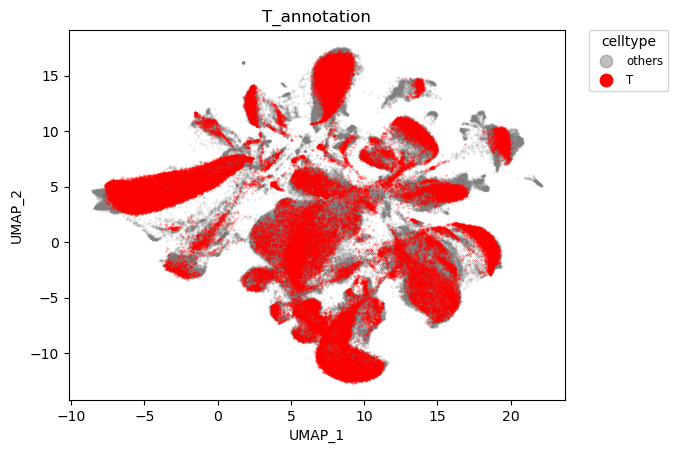
脑区对齐效果:
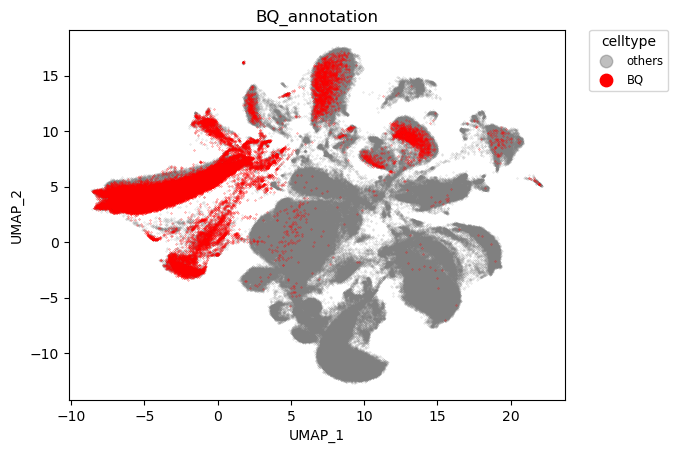
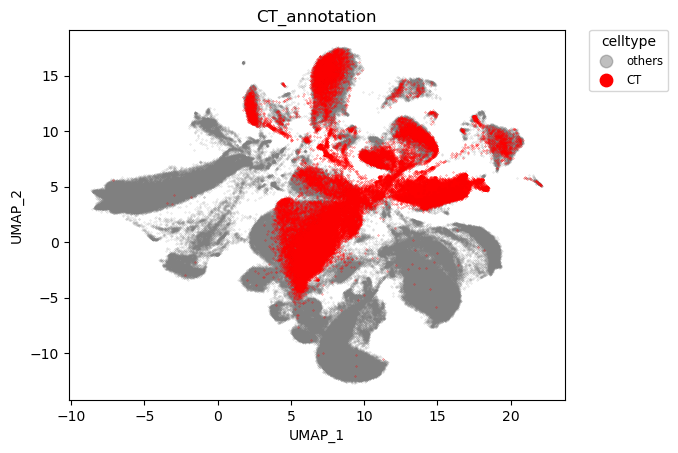
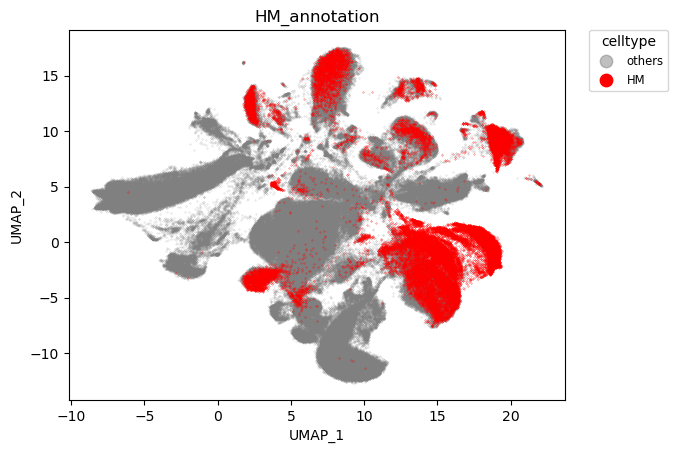
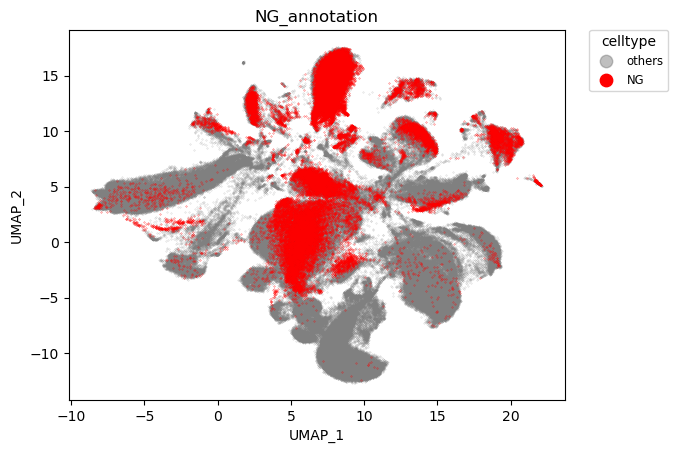
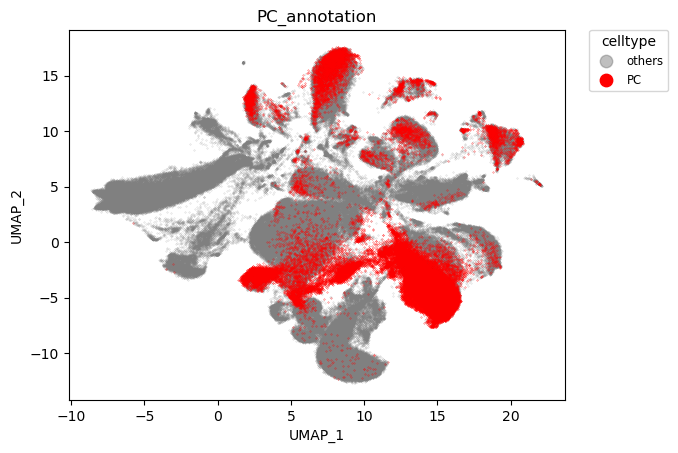
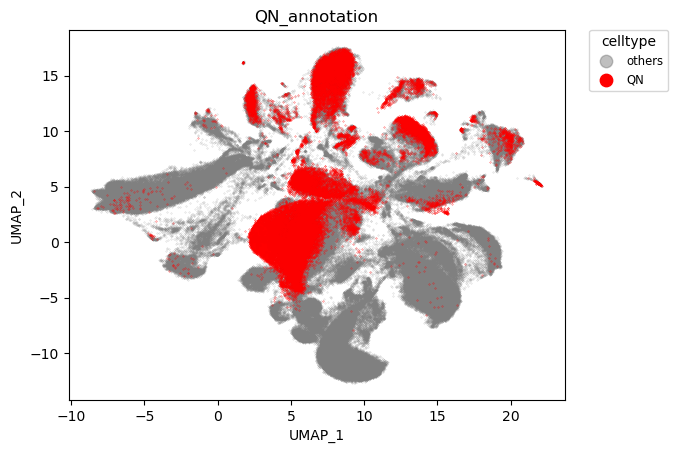
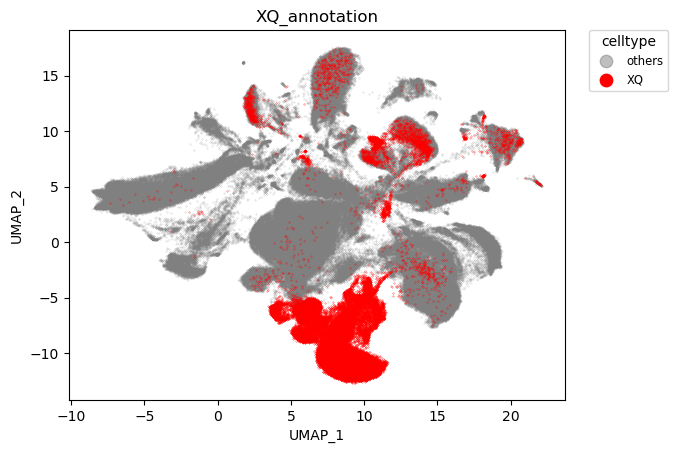
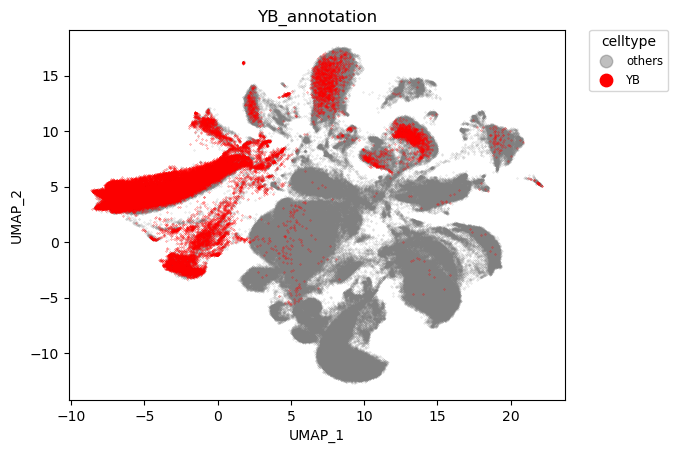
以上是关于单细胞数据整合的说明。
细胞类型注释
细胞类型注释通常有两类做法,基于marker gene的人工标注和基于参考数据组的自动化注释。这两类做法都有尝试。
人工标注的难点在于确定marker gene。文献中的一些marker gene会存在特异性不高(尤其是比较细的分类粒度,可能多个亚群都有表达)以及泛化性较差(即使是CNS上的工作,一些marker 也无法很好的迁移使用),而数据库中的一些marker gene则要考虑其可信度,CNS背书的marker gene可靠性会更高一些。
另外一个难点则在于判定的规则。有时候会不可避免地面临同一个cluster中多个细胞类型的marker gene同时高表达的情况或者所有marker gene 都低表达的情况。这时就很棘手。
使用自动化注释的方法确实可以回避人工标注的两个难点,但是同样有新的问题,一方面仍旧需要提供可解释的注释依据,另一方面则是无法有效的迁移。后面会详细解释这一点。
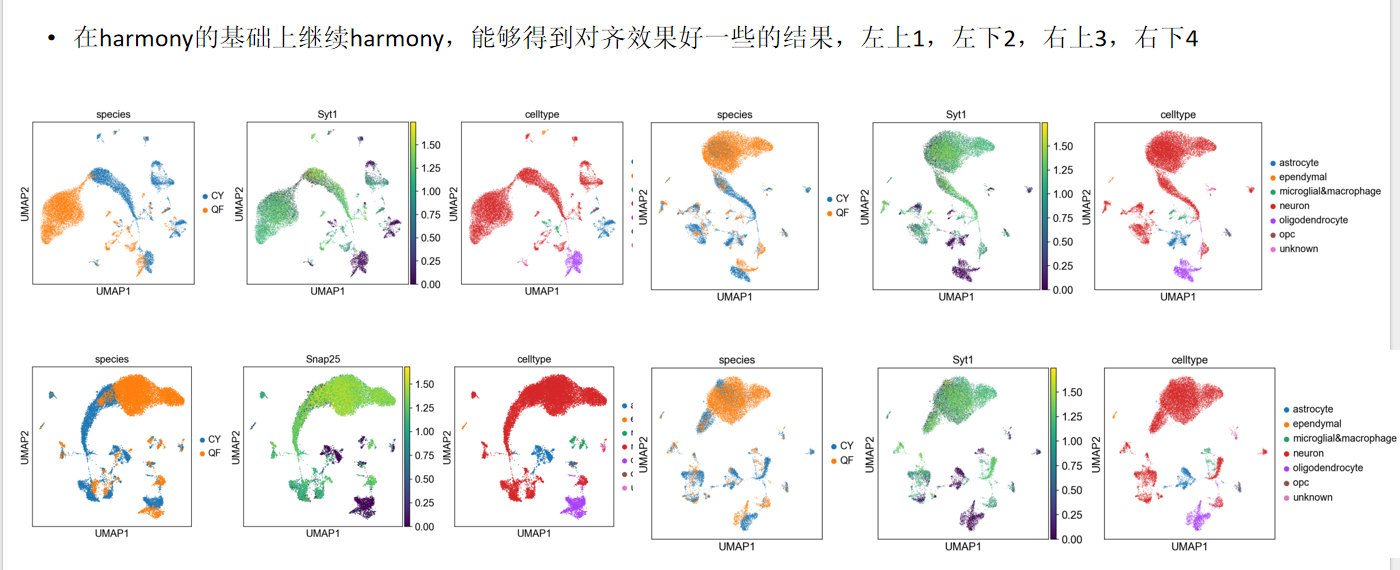
我最终选择的是人工标注。对于marker gene,我使用的是CellMarker 数据库加文献参考的方式扩充marker gene list。对于判定规则,除了常规的特异性表达,我也参考了2023年MIT的一篇工作2,如下图所示,可以看到研究人员在神经细胞亚型的注释上引入了且或的逻辑判定规则,这类判定对前面提到的多种marker gene的高表达的情形提供了解决的方案。

细胞类型判定规则
另外对于特异性表达这个概念的判定,也要根据数据质量进行适当的松弛。以24年这篇解读蝙蝠超声机制的工作为例3,可以看到其marker gene的表达百分比并不高,像astrocyte, endothelial, microglia这些细胞,可能20%-40%的一个表达比例就敢做注释,所以不要太过纠结dotplot上某个基因的表达百分比,可以和UMAP结合一下,有的表达比例不高的marker可能在UMAP上看起来效果还不错。
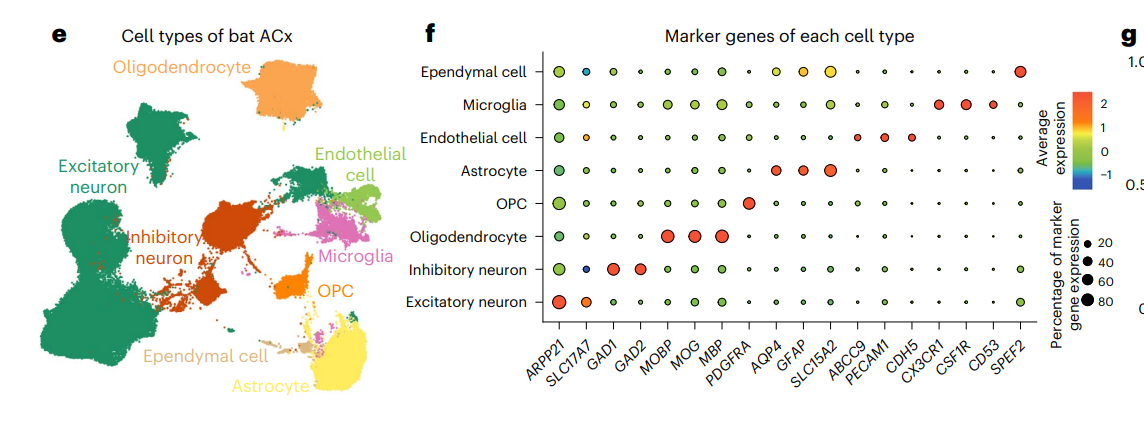
蝙蝠大脑单细胞注释
参考这些工作,最终得到的细胞注释结果如下:
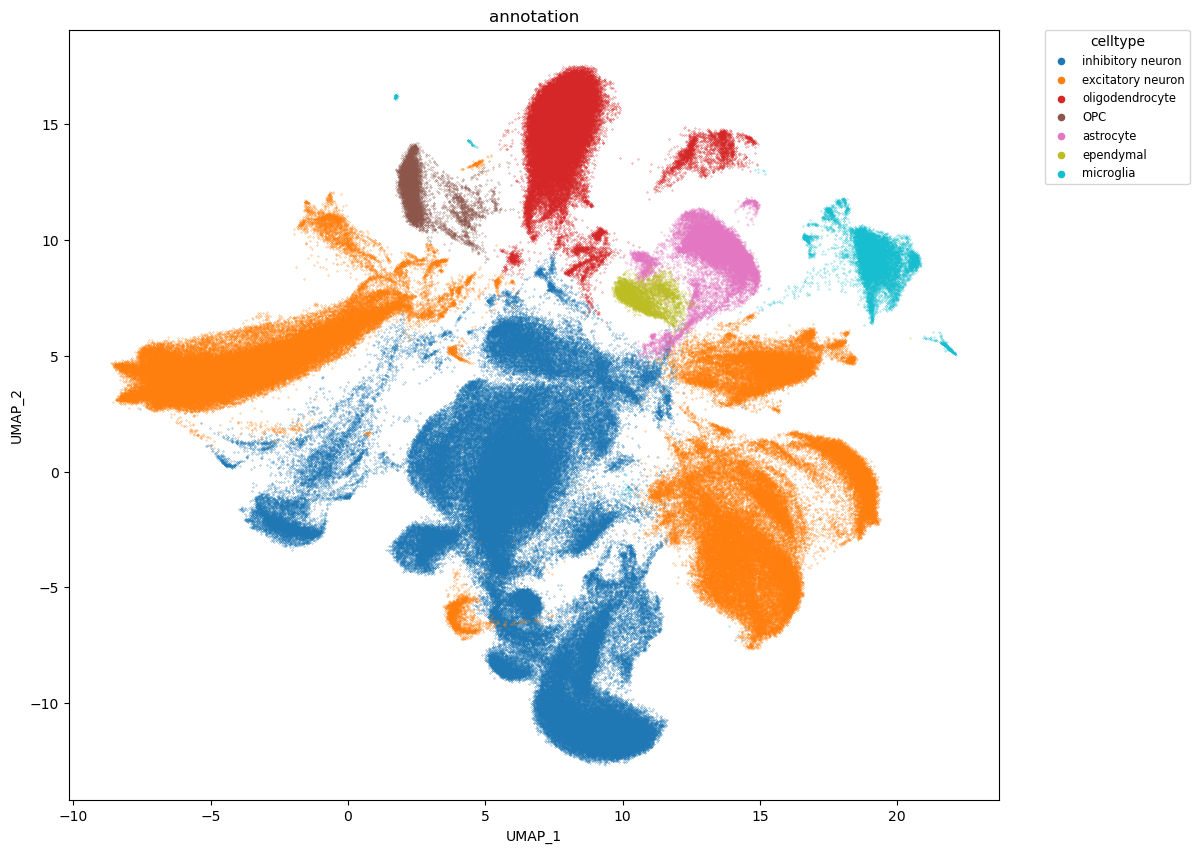
蝙蝠大脑单细胞注释
注释的依据如下,结合UMAP中marker gene的可视化、cluster层次的dotplot以及细胞类型层次的dotplot,可以相信这是一个合理的注释。
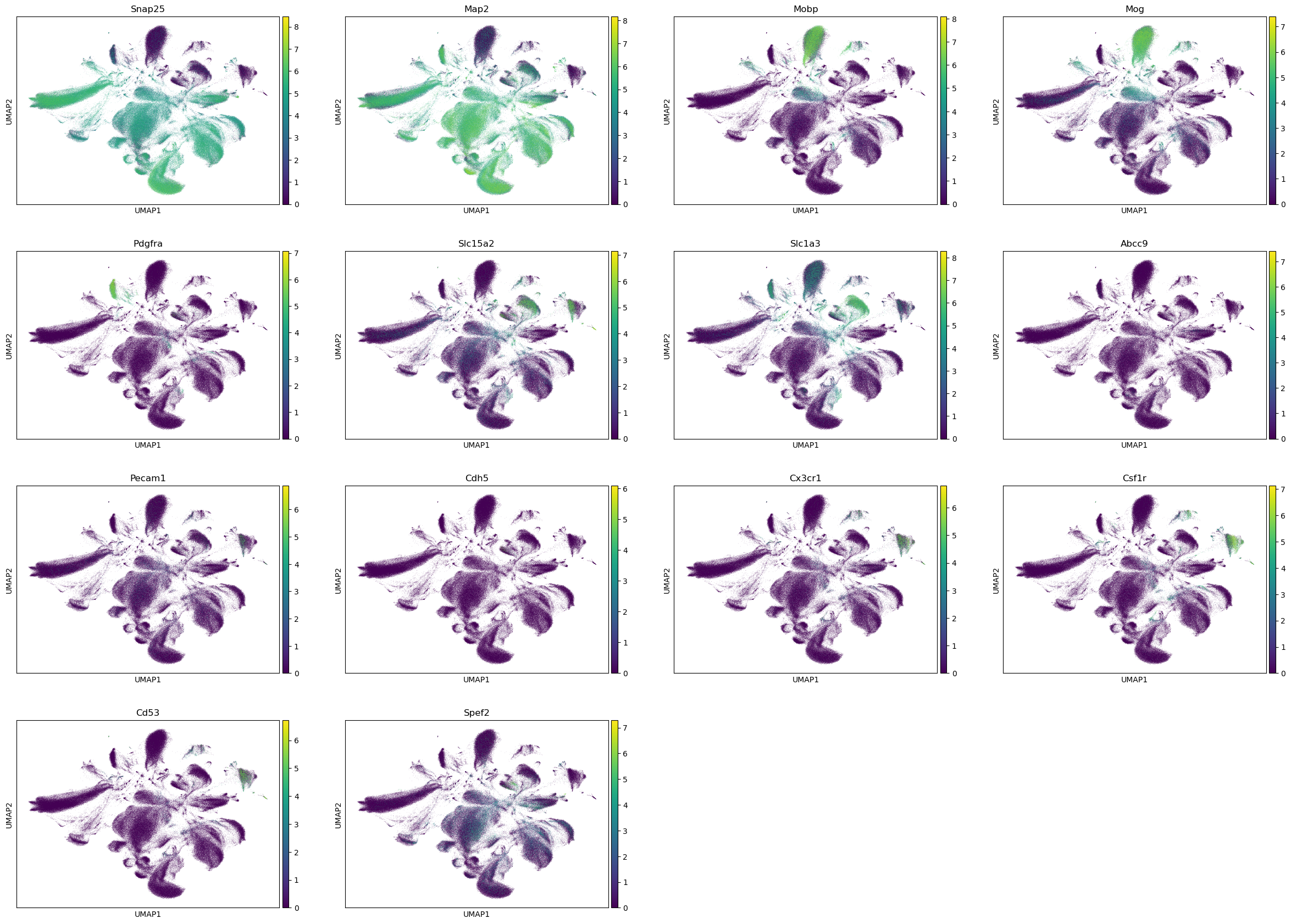
marker gene可视化
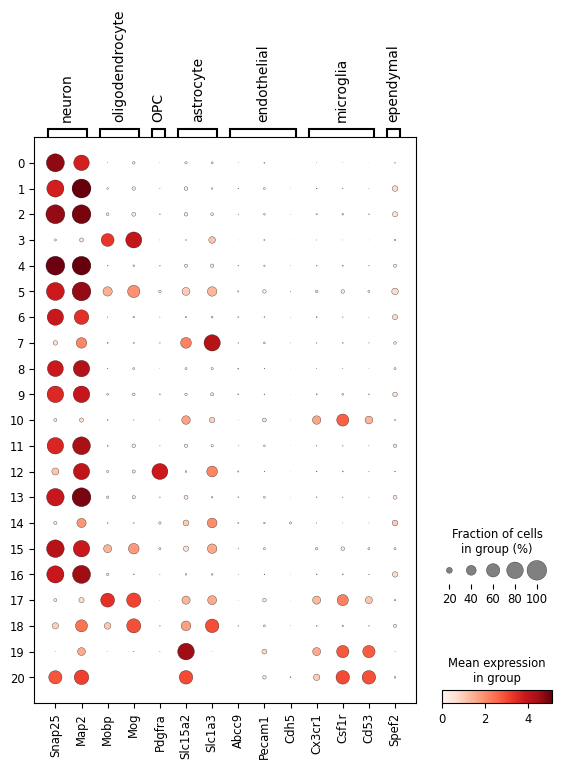
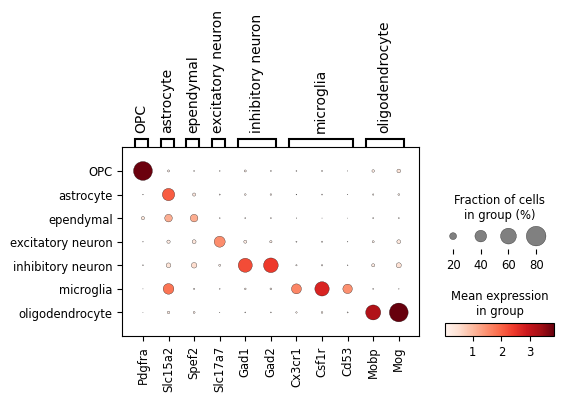
对于UMAP中一些细胞类型混杂的部分,我认为这是合理的现象,是忠于数据本身的分析的体现,可以参考1 3 中的UMAP结果,都是存在一定程度的混杂的。
总结一下细胞注释的基本方法:
- 结合研究问题和文献确定要注释的细胞类型,之后确定相应的marker gene list(在未界定数据集中包含的细胞类型的情况下,根据cluster的差异表达基因去匹配细胞类型是一种低效的工作方式,很容易匹配不上)。
- 结合UMAP,dotplot确定特异性表达并进行注释,不要太过纠结表达比例。
- 由粗到细的注释,逐级切分。例如先注释神经元非神经元,再在神经元下注释兴奋神经元和抑制神经元,非神经元下注释astrocyte, oligodendrocyte等等。通过逐级切分,控制背景信号。比如有的marker在全部细胞中看起来特异性一般,但限制在神经元中就可以良好的区分细胞亚型。
- 修正corner case。对不合逻辑的少量注释,或者删掉或者修正。
基于细胞注释,可视化细胞比例

细胞比例可视化
从细胞比例中,可以看到一个可能与回声定位相关的点,在海马中,两个非回声物种非神经元比例都有所上升。
曲折的试错经过
这里按照时间记录自己的试错经过。虽然这些是失败的结果,但同时也是教训,并且一定程度上显示了当前分析工具上存在的缺陷。
-
2023.09-2023.10
这一阶段主要是摸索单细胞数据分析流程,在小鼠海马和猪尾鼠海马上摸索demo。
-
2023.10-2023.12
这一阶段在小鼠海马和猪尾鼠海马上尝试了许多单细胞数据整合方法,当时感觉做不下去的点是,怎样算一个合理的对齐,以及对齐完之后又能做什么。
现在回看,这两点都可以被解决。合理的对齐来自于生物先验,缺少对相关文献的调研,导致将最开始使用mofa+的不充分对齐结果当作生物学差异,诱导出来错误的判断,同时也导致后期执着于对齐脑区差异,花费了大量的时间并最终失败。
而对齐能提供什么?在我看来真的没有很多,它可以帮你画一张漂亮的UMAP,告诉大家数据长这样。之后帮助你做细胞注释,在这个公共的细胞嵌入下,不同物种的相同细胞被聚合到一个cluster里,根据marker gene完成全样本的注释,不用一个样本一个样本单独的做过去。然后就没有了,因为对齐或者直接根据细胞间的knn图或者将细胞做了一个表示学习,两者都没有什么直接的生物解释性。所以真的做不了啥。
我个人认为,对齐就是承担一个提供宏观的数据分布和细胞注释的功能。这两步能很好的做完,对齐就已经发挥完其全部功能了。接下来重要的是差异比对,比如说同样是神经元,回声非回声的差异基因有哪些,调控网络有没有差异。甚至说,如果不需要数据可视化的话,不做对齐也无所谓,单个样本做注释、推断网络、推断细胞互作,之后做差异分析即可。
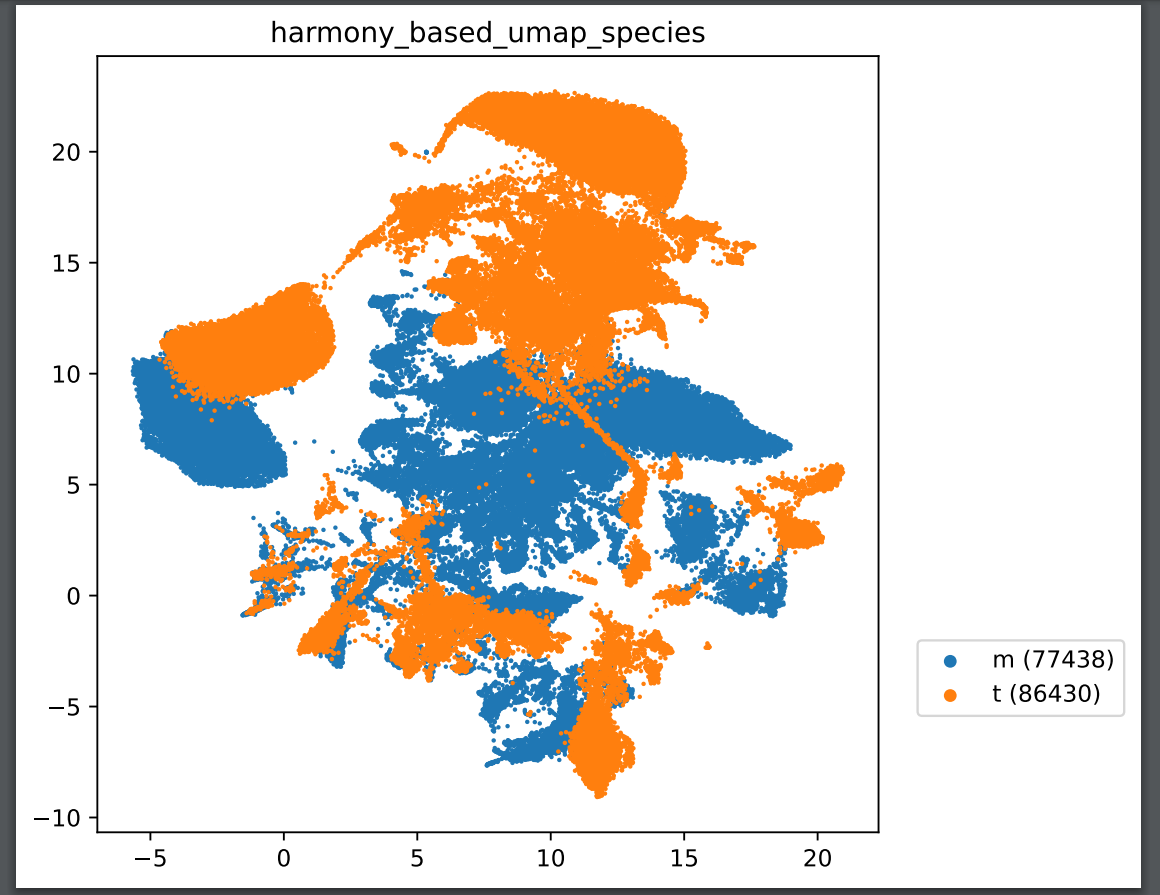
这一阶段我也暴露了一些其他的问题,例如夸大算法的影响。以对齐为例,harmony这种比较老的方法经常被一些新方法吊锤,这种偏见导致我对比整合方法时,看到Harmony表现较差就直接归因于方法落后而不是自己调参失败或者对齐的因子不对。结合我最后使用harmony对齐的效果来看,在选好对齐的因子为物种时,harmony在5个物种的对齐上做的很好,在2个物种的对齐上表现较差(如下图所示),但是如果调整惩罚系数或者进行多步harmony,其在2个物种的对齐上也表现不错。因此不要过分迷信论文中算法的性能,结合数据好好调参更有效。
-
2023.12-2024.03
这一阶段的主要工作是参考2023年发表在NEE上的一个工作6,去实现自己数据的分析。虽然两者研究的问题很相似,但是直接迁移的结果表现并不好,最终也被放弃。
当初选择NEE流程的原因是,其回避了精细的细胞类型注释和数据整合这两个我当时未很好解决的问题,仅将灵长类的大脑细胞注释为兴奋神经元、抑制神经元、非神经元三大类。在细胞类型的差异分析上,这篇工作对每个物种单独做聚类,之后应用metaneighbor7这个方法比对各个cluster之间的相似程度,将这些物种特异的cluster进行聚合,从而区分物种特异和物种保守的cluster。
但在实际应用中,发现在NEE的聚类粒度下,我们的数据得到的聚类结果基本上是不可信的,一方面是物种保守的cluster占比很少,另一方面这种物种保守的cluster中混杂了神经元和非神经元,难以进行后续分析。如下图所示:
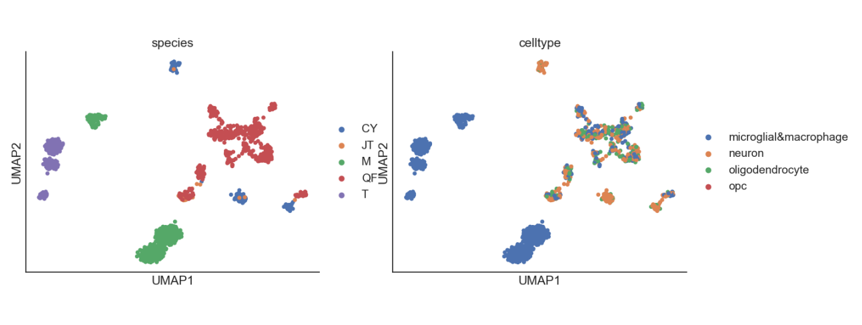
失败的meta-neighbor分析结果可以看到虽然这个cluster是保守的,五个物种都有,但是里面的细胞类型是杂乱无章的。
现在回看这部分,太细的聚类粒度导致细胞类型混杂是主要问题,如果在当前的细胞注释下,对每个细胞类型进行这样的精细聚类,之后再进行merge,可能可以解决这个问题。至于当时为什么不采用这种做法,原因是细胞注释不够精细,当时无法区分兴奋神经元和抑制神经元,而这个区分在我们看来是必须要做到的,做不到这一点的细胞注释结果意义不大。
当然这部分的尝试并不是毫无意义的,meta-neighbor这个方法给了我们一个量化数据集差异的方法,后面我们用这个方法确认海马是八个脑区中回声非回声差异最大的部分,为后续我们针对海马深入分析提供了支持。

meta-neighbor发现海马在回声-非回声物种中的差异最明显 -
2024.03-2024.05
在意识到必须要注释出兴奋神经元和抑制神经元这点后,这一阶段围绕这个目标开展,尝试了40个样本逐样本的单独人工注释和使用参考数据集的自动化注释。
人工注释由于当时自己过于严苛的标准导致未成功注释,后续转而使用小鼠大脑的atlas数据集进行自动化注释。前期使用21年的工作进行注释8,总体表现还不错,计算cluster之间的cosine相似度就能实现不错的注释效果。但后面考虑到这组数据只涉及到小鼠的海马和垂体两个脑区,用来注释全部的8个脑区可能会导致问题,因此改用23年的全脑数据注释1,结果出现了逆天的效果:
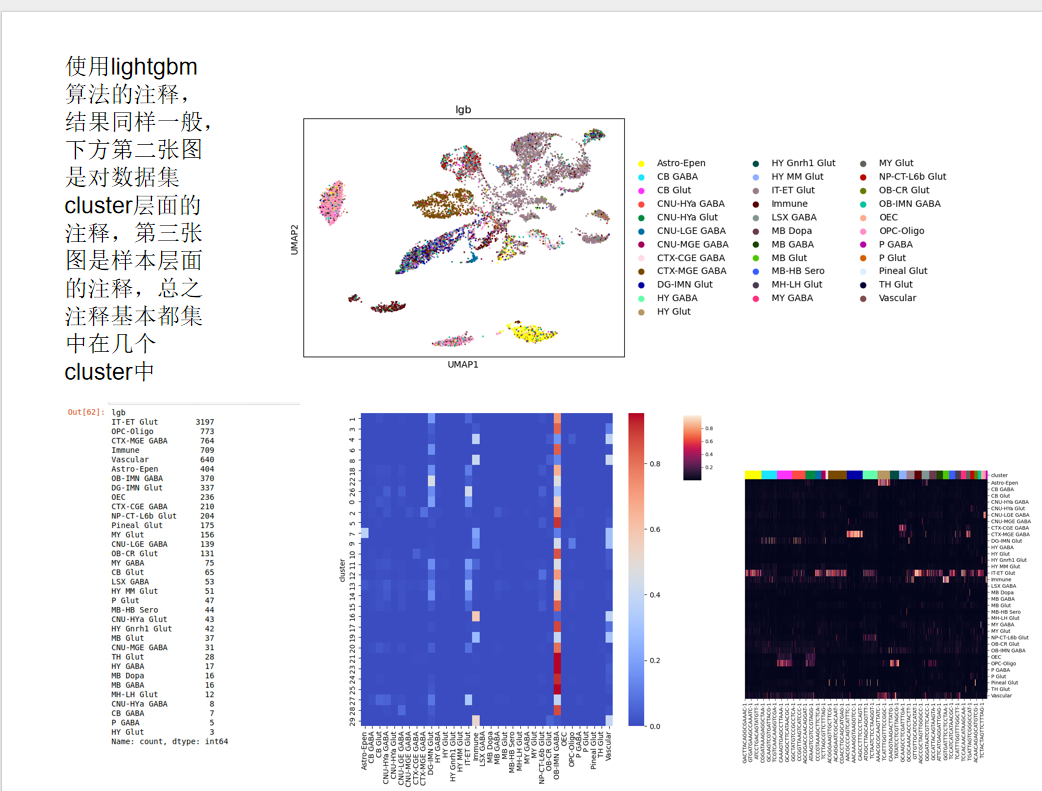
不理想的细胞注释结果,细胞类型混杂严重感觉出现问题的点可能是单细胞的测序深度问题,23年的数据测序深度明显比21年和自己的数据深,测序深度对数据整合以及标签迁移的影响不能简单通过一个CPM标准化实现,将特征转成rank特征、消除量纲的影响也无法改善这一点。
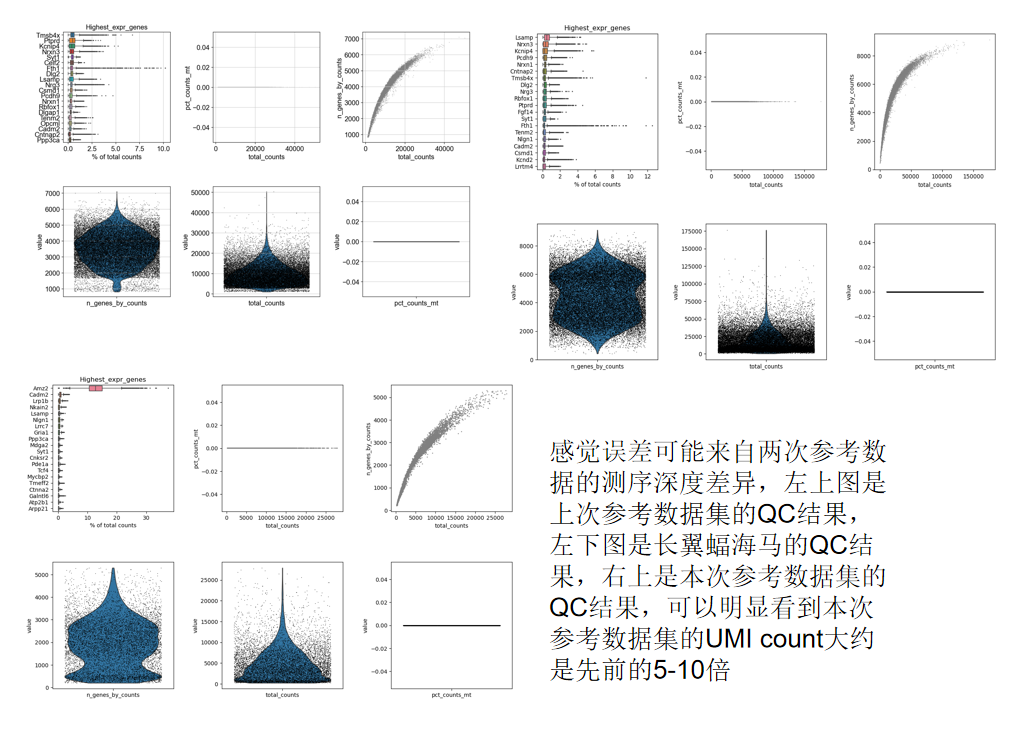
测序深度差异 -
2024.06-2024.07
这一阶段看到了这篇论文的注释策略3,发现只要恰当松弛注释标准后即可完成兴奋神经元和抑制神经元注释,同时再次研读了23年工作的整合3,发现脑区是很难对齐的。在这两篇工作的指导下,迅速完成数据整合和细胞类型注释,并且进行后续的调控网络推断以及差异分析步骤。
scATAC-seq数据处理及调控网络推断
scATAC-seq数据处理
对于scATAC-seq数据,主要是将其应用于调控网络的推断。这里解释一下这类数据的基本格式。
前面提到,scATAC-seq是将DNA序列片段和基因组序列进行比对,因此对每个细胞来讲,ATAC-seq数据可以看作是一个空间序列,空间信息就是其在染色体上的位置,如果某个片段被Tn5转座酶切割下来的话,对应的区间值为1,否则为0。
一个示意图如下:
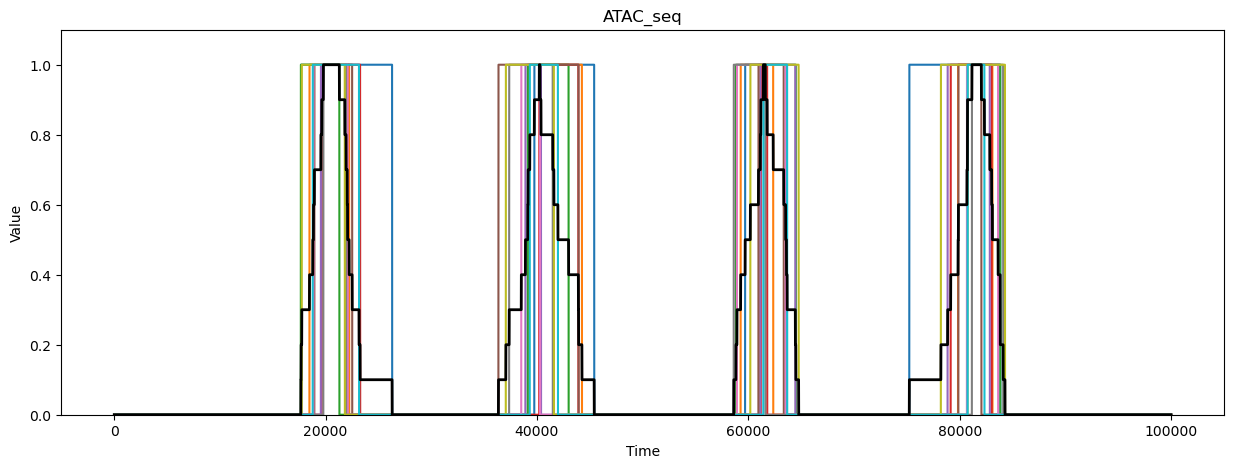
其中彩色的线代表每个细胞的atac信息,可以看到这些细胞在20000,40000,60000,80000这四个位点附近有片段被切割(value=1)。但由于每个细胞切除的片段大概率不一致,所以距离atac-seq数据分析常见的cell-peak矩阵依旧存在一定的距离。这时需要一个call-peak的过程,即将这些片段富集的区域做为一个peak,图中的黑色折线是一个不严谨的示意图,我们可以根据黑色折线中高于阈值的片段确定peak,之后统计每个细胞在这个peak区间里的片段数目。
注意,虽然每个细胞中得到的片段数目至多为1,但经过call-peak后,peak count的值是一个大于等于1的整数,示意图如下:
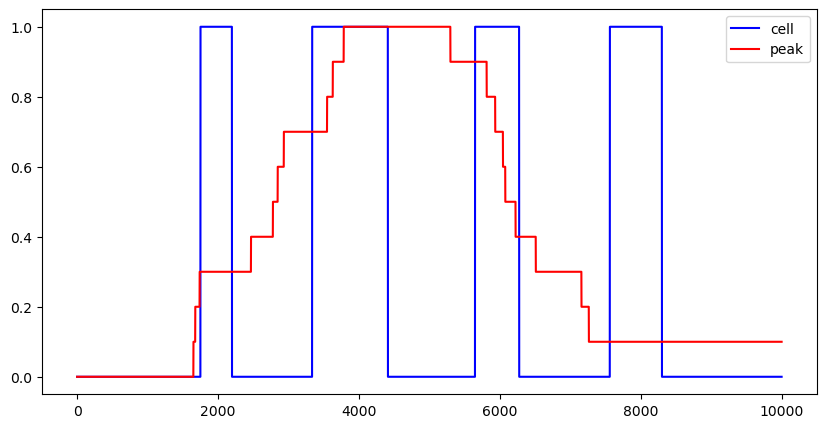
可以看到在红色折线对应的这段peak区域,这个细胞可以被Tn5切割成多个片段,因此在最终的cell-peak矩阵中,该细胞在该peak处的值应该为4。
以上是scATAC-seq数据的基本格式,这里主要想解释一下为什么cell_peak矩阵中有大于1的值,当初寻思了一会这个问题。
对于scATAC-seq数据后续的分析,由于我并未展开相关工作,暂时跳过该部分。
调控网络推断
基因调控网络(GRN)通常有两种类型,根据网络中边的关系,可以分为TF-TG网络 和 TF-RE-TG网络这两类。其结构应当是一个稀疏的有向图,稀疏性是因为调控下游基因的转录因子,不太可能是全部的几万个基因。
比起scRNA-seq数据,ATAC-seq数据给我们提供了peak(RE,调控元件)的信息,这会帮助我们建立RE-TG 和 TF-RE之间的关系,因此可以通过ATAC-seq数据更好的建立符合调控机制的TF-TG关系。
对于调控网络推断,我选择的是celloracle方法4 ,主要原因是原理简单,流程容易跑通。其主要步骤概括如下:
-
筛选peak-tg 之间关系,这一步主要目的是降维和保留有意义的调控关系。通常根据两个阈值来筛选,第一是距离,不在同一条染色体上的peak和tg、以及距离过远的peak和tf,大概率无调控关系,删掉。peak和tg之间相关性过低的,大概率无调控关系,删掉。
怎样构建peak和tg之间的相关性呢?可以直接计算peak和tg之间的相关系数,也可以计算peak和该基因转录起始位点周边peak之间的相关系数,有专门针对peak相关性的计算工具cirero5,得到peak-peak之间的相关性矩阵,再根据peak_annotation文件得到peak-gene之间的相关性。
如果使用10X多组学工具的话,peak相关性和peak_annotation文件都被提前计算好了。只需要 将这些信息提取出来即可,可以参考multivelo中的aggregate_peaks_10x函数进行简单修改。
-
筛选peak-tf 之间关系,这一步是利用peak对应的序列信息和tf对应的motif信息计算motif结合分数,从而筛选和该Peak区域结合的tf。使用Homer这类motif 扫描软件实现。对于非模式物种,需要提供原始的基因组序列数据,例如.fasta文件。
-
最后一步是建立tf-tg之间关系,这样的tf-tg关系也被记作base_grn。将有公共peaks的tf,tg建立这样的关系。
-
基于base_grn,应用celloracle进行网络推断。
小结一下,ATAC-seq数据分析的难点主要在于其数据并不像scRNA-seq那样结构化。需要一些额外的结构化操作。对于调控网络的推断,难点主要在于繁琐的前处理过程。
Atlas数据分析的流程化处理
本次分析的单细胞数据量级较大,共有40万个细胞,并且样本中没有重复样本,差不多也算是一个atlas量级的数据了。由于样本量较多,不可能像几个样本的情况下那样逐样本精细化分析,必须建立自动化的分析流程。快速得到全局的信息,之后定位到局部(海马)进行精细化分析。
对于流程搭建,我认为需要关注以下几点:
- 数据结构化,在前期将不同类型的样本数据尽可能统一成结构化的数据格式。
- 程序模块化,脚本复用,保证能够快速迭代。
- 写好日志文件,保留关键步骤信息,方便出问题后纠错。
- 先搭好baseline。不要纠结于某些计算工具的选型,在合理的前提下选最常用的、最容易跑通的而不要选所谓性能最好的。在全局计算的层面上,计算工具的影响没那么大。
另外就是研究目标的“抓大放小”,因为数据量大,40个样本随便组合下都能看到很多有意思的信号,要关注最有可能有进展的,其他的信号可以记下来后面看。全局信息的获取可以粗放一点,40万个细胞扔掉几万个质量差的没有问题。
最后还有数据的及时备份,6月份管理员误删我原始数据差点给我干没……
后续研究思路
对于这个课题来讲,后续研究主要集中在差异分析上,建立调控差异、基因表达差异和回声定位性状的关系。
除了上述的分析工作,在做先前的分析工作时,遇到的一些问题也提供了后续方法改进的空间。主要有以下几点:
-
自动化细胞类型注释加生物学解释性。如果测序深度的影响验证为真,可以针对这一点进行改进。同时从用户体验来讲,提供像2中基于且或这样的判别依据,一方面可能比单纯的marker gene能更准确的注释,另一方面也有助于用户迅速验证自动化注释的合理性。
-
多组学多样本的数据整合。
-
端到端的网络推断,不同的多组学数据对应不同的peak信息,尝试设计一个peak编码器,能够直接根据位置信息加基因组序列数据给出初始编码,这样能够减轻预处理部分的难度。同时通过和细胞嵌入进行耦合恢复表达信息,实现 in-context peak embedding,用于后续的网络推断。
参考文献
- [1] Yao, Zizhen, et al. “A high-resolution transcriptomic and spatial atlas of cell types in the whole mouse brain.” Nature 624.7991 (2023): 317-332.
- [2] Langlieb, Jonah, et al. “The molecular cytoarchitecture of the adult mouse brain.” Nature 624.7991 (2023): 333-342.
- [3] Liu, Meiling, et al. “Complexin-1 enhances ultrasound neurotransmission in the mammalian auditory pathway.” Nature Genetics (2024): 1-13.
- [4] Kamimoto, Kenji, et al. “Dissecting cell identity via network inference and in silico gene perturbation.” Nature 614.7949 (2023): 742-751.
- [5] Pliner, Hannah A., et al. “Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data.” Molecular cell 71.5 (2018): 858-871.
- [6] Suresh, Hamsini, et al. “Comparative single-cell transcriptomic analysis of primate brains highlights human-specific regulatory evolution.” Nature Ecology & Evolution 7.11 (2023): 1930-1943.
- [7] Crow, Megan, et al. “Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor.” Nature communications 9.1 (2018): 884.
- [8] Yao, Zizhen, et al. “A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation.” Cell 184.12 (2021): 3222-3241.