最近想借助transformer架构去解决手头数据的建模分析问题,同时,本着“击败敌人首先要了解敌人”的思想,想要证实我的一些大逆不道想法,还是要深入了解一下scRNA-seq预训练模型这一两年来的进展。这篇博客是对近期的一些论文阅读的总结。
首先感谢OmicsML ( https://github.com/OmicsML/awesome-foundation-model-single-cell-papers )这个开源项目,将这几年来的一些预训练模型(foundation model)做了一个详尽的收录,我也是参考里面的论文列表去阅读的。下面将尽可能按照时间顺序去总结一下这些工作。
预训练模型
scFormer(2022.11.22 biorxiv)
这篇论文是wangbo团队2023年ICLR的一篇投稿论文,2022.11.09被拒后就挂在了biorxiv上。看了一下,应该是后面scgpt的初代版本,框架上还是一个encoder-only的架构,没有修改成后续scgpt的生成式模型。实验验证部分也比后面scgpt少了许多。因此这里不再对文章进行分析。值得关注的是文章的openreview,很多审稿人的意见很有价值。这里总结一下:
- 对非序列数据应用transformer架构的动机。(个人觉得把图片做成patch然后transformer也没多少序列结构信息,这个意见我不是很赞成)
- 对gene embedding的生物学意义没有做深入分析。(同意这点,transformer结构里的embedding含义是值得分析的)
- 评估时比较的方法不充分。(同意,尤其是在细胞任务上,应该与MLP,VAE这种框架的同等大小的模型比对)
- 掩码预测损失使用cross-entropy。(同意,将一个基因表达量为5的基因预测成4和预测成0的误差显然是不一样的,cross-entropy没有考虑这一点)
- 新颖度不够,和scBert太像了。(同意,只是loss部分的修改)
综合上面的意见,下游的评估和非序列数据应用transformer架构的意义是scRNA-seq预训练模型中的核心问题。对于后者,把transformer看成是一个高次的编码器而不是一个序列数据上的模型或许会好些?
scGPT(2024.02.26 nature methods)
参见 https://moonbyul-99.github.io/2024/03/30/scGPT%E8%AE%BA%E6%96%87%E8%A7%A3%E8%AF%BB/
SATURN(2024。02.16 nature methods)
参见 https://moonbyul-99.github.io/2024/02/19/SATURN%E8%AE%BA%E6%96%87%E8%A7%A3%E8%AF%BB/
scHyena(2024 biorxiv)
韩国KAIST团队2024ICLR的投稿论文,被拒。看下来,仅是将全新的模型结构Hyena应用到单细胞数据上,支持全长基因的训练。主要卖点就是这个Hyena架构的full-length,没有仔细研究这个架构,貌似是牺牲了一些self-attention的全局交互特性,在序列长度上取得了一些进展。后面的下游任务做的挺糙的,就一个数据填补和细胞类型注释。
审稿意见如下:
- 下游任务太少太简单,基线模型过时。dropout问题没有显式的解决。
- 数据量是否充足?仅有400k数据。
- 消融实验不充分。
总结下来就是工作量并不让人信服。将一个架构应用到scRNA-seq数据上并不足以支撑这份工作,需要更全面的、更有挑战性的评估任务来说明新架构优势。
cellPLM (ICLR 2024 poster)
cellPLM在模型架构和分词上都做了调整。先前的绝大多数工作都是将一个细胞的基因表达看作是一个句子,根据细胞的部分基因表达预测掩盖住的基因表达,cellPLM则是将一个细胞看作是一个单词,一个batch的细胞看作是一个句子,在整个batch下进行掩码预测。
模型框架如下:
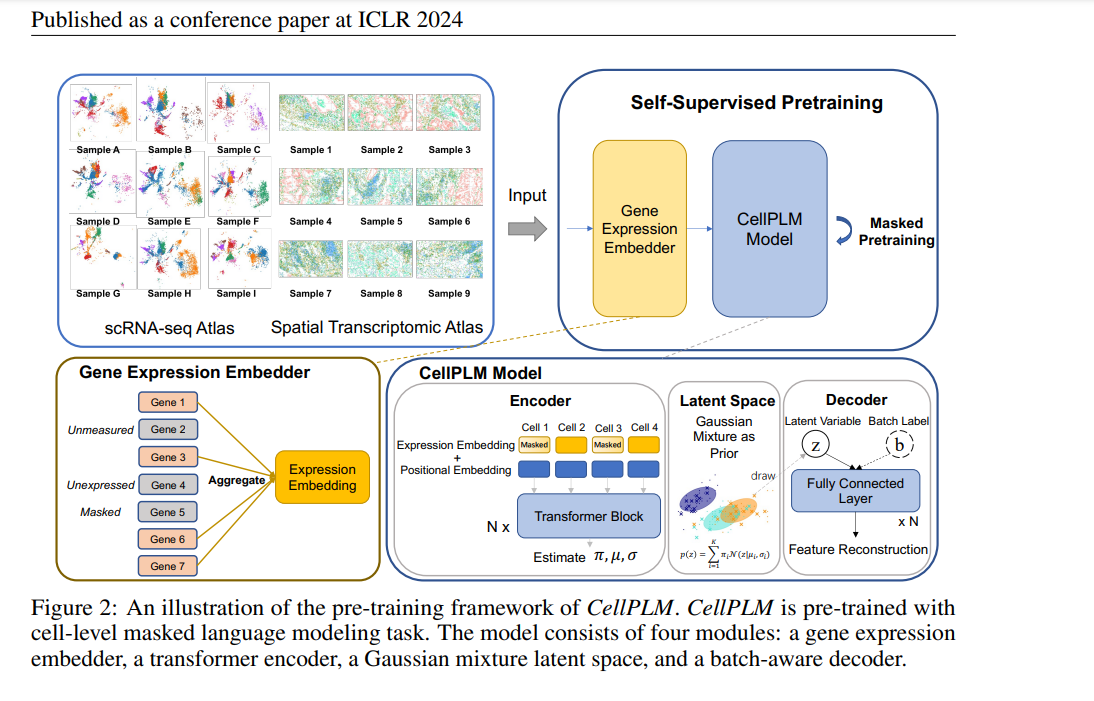
基本计算步骤如下:
输入的batch数据 $X \in R^{B\times p}$, (经过标准化和log变换的数据), 掩码矩阵 $M \in {0,1}^{B\times p}$
- gene expression embedder: $E = XW \in R^{B \times k}$
- 之后在transformer encoder中得到 $H^{(l)} = TransformerLayer^{(l)}(H^{(l-1)}) \in R^{B \times k}$, $H^{l}$的每一行代表一个细胞的嵌入。
- Gaussian mixture latent space,没有太看明白,我的理解是要使得嵌入向量能够反映这个batch中的不同cell cluster信息,因此对一个gaussian mixture模型进行优化。
- MLP decoder,将$H^{l}$从$R^{B\times k}$映射到$R^{B\times p}$作为对原始的基因表达矩阵的重构,使用MSE loss。
训练数据上,使用了9M scRNA-seq数据和2M 空转数据。
个人感觉cellPLM还是很有新意的工作,将cell看作单词而不是句子极大的提高了数据的处理效率。整个数据集可以在一个前向传播后就拿到结果。
scCLIP (NeurIPS2023-AI4Science Poster)
单细胞多模态预训练模型,clip的直接应用。模型框架如下:
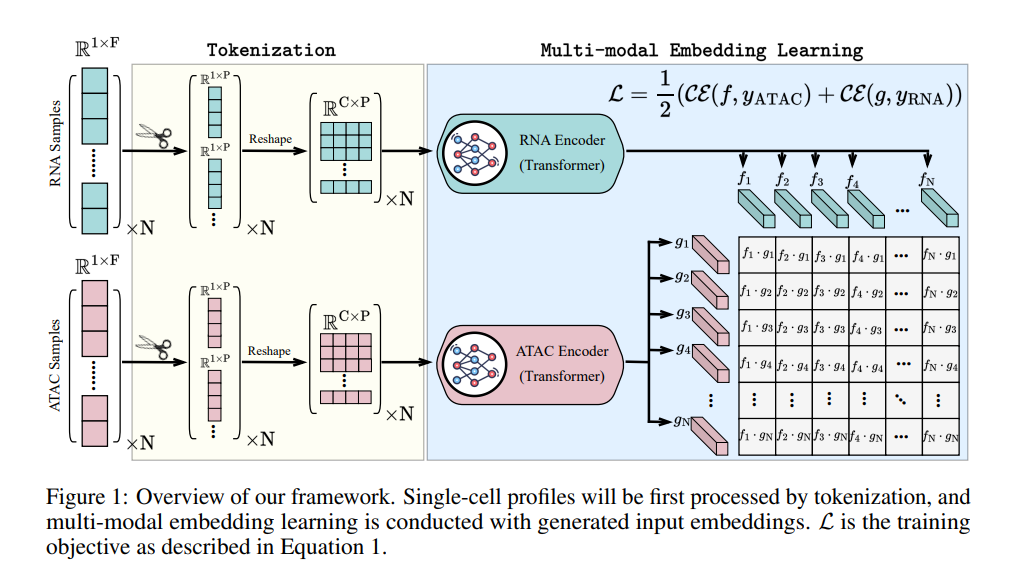
有点类似于图片的patch处理,直接将基因表达向量、染色质开放向量 reshape中一个(c,p)大小的矩阵,然后套clip框架。后面的评估任务也很局限,就只能做个整合和聚类。
用的数据量也不大,极大的鼓励了我去训练一个自己的预训练模型。另外,这个框架已经局限在处理细胞层面的任务了,没有得到一个基因或者peak的embedding,后面很难做调控相关的任务了,做不了调控的多组学框架没前途。
UCE (biorxiv 2023.11.28)
常规的框架,嵌入部分用了染色体位置信息和ESM2嵌入,别的改动不大。就是做的大,650M参数。文章题目给的很大:Universal Cell Embeddings: A Foundation Model for Cell Biology。
号称使用ESM2嵌入就能够实现跨物种整合,如果拿不到物种的蛋白质序列信息呢?拿个DNA语言模型做嵌入应该普适性更高吧?Universal存疑。
Nicheformer (2024.04.15 biorxiv)
Fabian J.Theis组的单细胞+空转大模型,比cellPLM做的要大,评估也做的更多,其他没啥印象了,有点让人失望,我对Theis组的期望一直蛮高的。空转信息似乎只在后面空转任务微调时用到。
scPRINT (2024.07.29 biorxiv)
一作是OmicsML项目的主要贡献者之一,老哥做了几年生信工程师后又开始读博,可以的,scPRINT的整个代码写的确实很漂亮。还是常规的transformer框架,卖点在基因互作网络上。基因互作网络还是基于注意力矩阵得到的,但是老哥加了一个集成机制来更好的处理多头注意力的多个注意力矩阵。另外,手绘的主图很震撼。

一个不同的点是预训练任务不是完全无监督的,包含有监督的细胞分类任务。受益于cellXgene数据库的需求,所有细胞都是有注释的,作者据此构建了一个一致的层次的细胞注释。花点时间做数据一定是有收益的。单细胞的预训练范式确实应该考虑下了,就算不愿意精细注释,做个pseudo-label做点有监督半监督的训练大概率比一直无监督的掩码训练要好。
预训练模型评估
预训练模型的评估有些被忽视,但是好的评估准则对做方法是非常有必要的,甚至比一些盲目堆数据堆参数小改一下架构的预训练模型更有价值。
Metric Mirages in Cell Embeddings (biorxiv, 2024.04.02)
通讯作者Aviv Regev,非常欣赏的工作。作者质疑了当前数据整合的主流评价标准,构建了一个模型,在主流评价指标下远超竞品,但是实际整合的结果并不具有生物学意义。
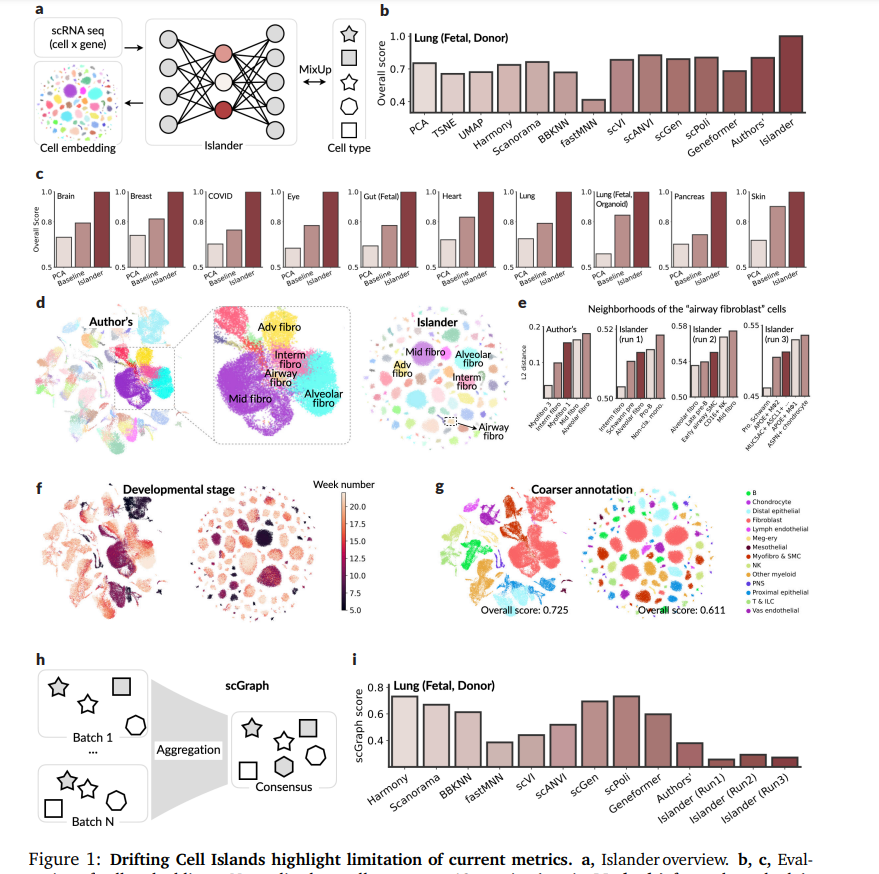
上面子图c显示了在不同组织下,作者训练的这个误导模型拿到了最高的分数,但是在d,f,g图中可以看到,一些原本关系很密切的细胞,在误导模型给出的嵌入下分散在各个位置,误导模型的嵌入丢失了原有的生物信息。
一个解决方法是作者提出的scGraph评价指标,简单说就是不只要考虑分类性,更要考虑其细胞谱系的特征,嵌入应该很好的保留细胞谱系的信息。这给当先众多做数据整合的工作提了一个醒。
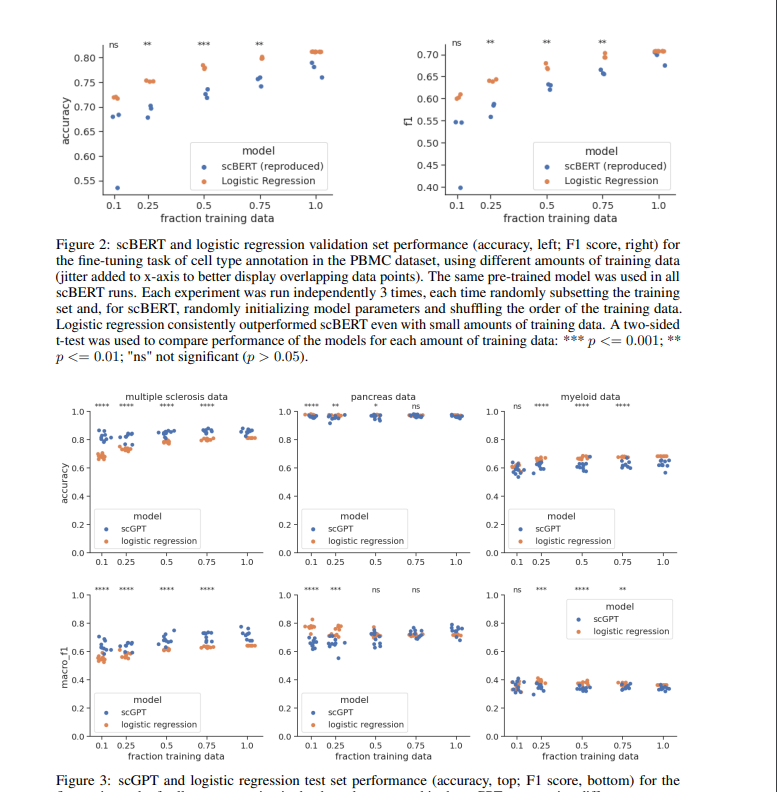
A Deep Dive into Single-Cell RNA Sequencing Foundation Models(biorxiv, 2023.10.23)
很有价值的工作。细胞注释一直是单细胞预训练模型的一个重要评价指标,这篇工作通过实验说明,简单的Logistic regression就足以达到预训练模型的精度了。因此,如果想要说明单细胞预训练模型的价值,别刷细胞分类了,so easy。
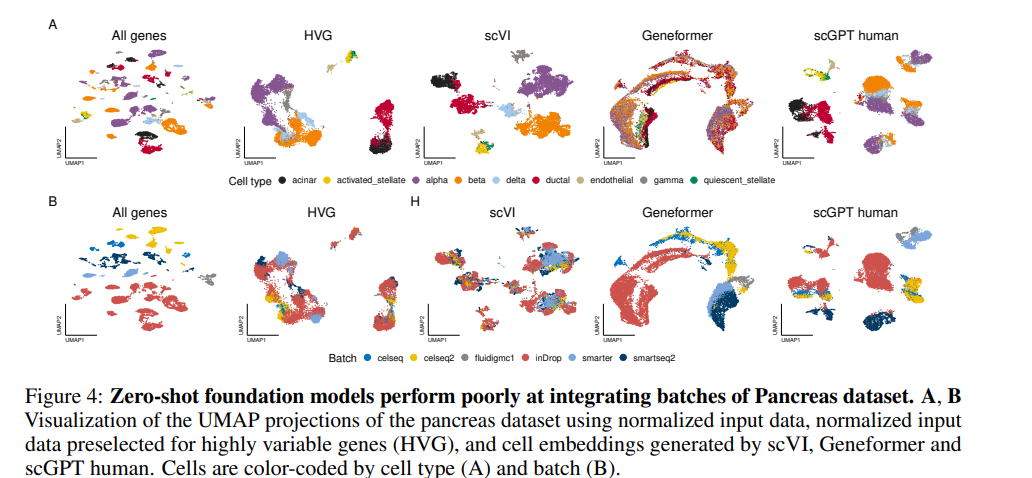
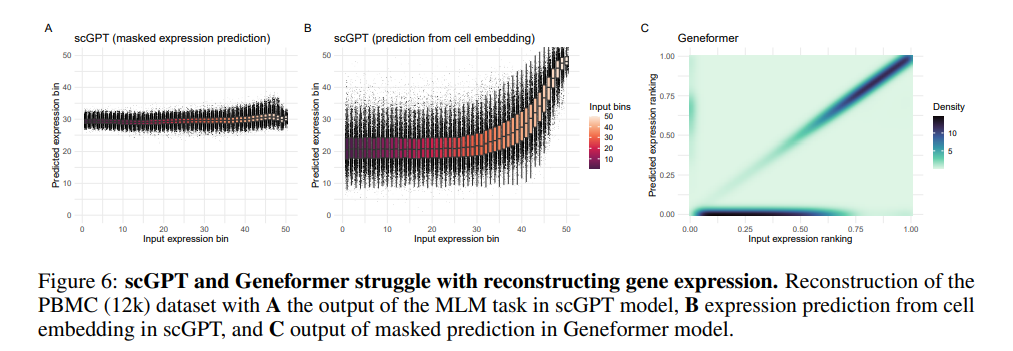
Assessing the limits of zero-shot foundation models in single-cell biology (biorxiv 2023.10.16)
文章主要质疑预训练模型以下几点,embedding的聚类结果很差,zero-shot根本没有达到文章宣称的能力,甚至连预训练的目标掩码预测的效果都很糟糕。好好想想到底训了个啥玩意儿吧……
Foundation Models Meet Imbalanced Single-Cell Data When Learning Cell Type Annotations (biorxiv 2023.10.24)
很糟糕的写作,没有什么内容,这个工作的评估做的很粗糙,不推荐读。
小结
模型部分,cellplm, scprint是更有意思的工作。评估部分,考虑细胞谱系的整合指标和更难的细胞层面任务是值得思考的方向。
单细胞预训练模型现在是一个挺卷的方向,都怪OpenAI,人人都想来蹭大模型的热点了,但就单细胞预训练模型这边,截至目前我认为还没有看到一个非常出彩的工作。因此,虽然卷,但是还有很多值得去做的内容,比如transformer是否唯一解,花点时间做更高质量的数据有多大收益,有没有比细胞聚类更靠谱的评价任务等等。最重要的是告诉生物研究者,花这么大成本做的所谓foundation model靠谱吗?不靠谱赶紧换个小模型玩吧……