下次组会准备讲 Multiomic foundation model predicts epigenetic regulation by zero-shot 这篇论文,这篇Blog记录一下自己对论文的一些想法。
简介
阿里达摩院团队的工作,提出了一个CREformer模型,可以用于基因调控的zero-shot推断。
看下来觉得主要亮点是多模态数据(DNA序列数据,组学数据), 多样性的组学数据(ATAC-seq, chip-seq, RNA-seq) 以及不同尺度的组学数据(bulk, single cell)的联合预训练。下游任务看起来还不错,让我对调控推断稍微恢复一点信心。
基本思路
在开始梳理模型架构和分析结果之前,先思考一下 why we need CREformer?
考虑一个调控关系,TF结合RE进而影响TG,在这个过程中的影响因素会有?
- 序列信息(DNA 序列,motif序列决定TF-RE的结合潜力,RE和TG之间的距离影响调控强度),显然序列信息是物种特异的,不足以解释一个生物体内组织、细胞类型之间的差异(同一个个体序列信息是固定的)
- 组学信息(ATAC-seq,RNA-seq), 这些信息是组织、细胞差异的信息,借助这些信息可以分析一些组织特异或者细胞特异的调控关系。
因此结合序列信息和组学信息的调控建模,应当能抓住不同粒度的调控关系(物种特异,组织特意,细胞特异)。
再考虑一个调控模型能带来什么?
- 调控关系推断
- 基于调控关系进行基因表达的扰动预测
模型细节
CREformer-Elementary
-
预训练数据:
使用ENCODE内的数据,34个样本,7种不同的表观信号,外加物种的DNA序列。对每个样本,从基因组序列上随机抽取$50K\times 32K$ bp的non-overlapped序列。最终有 $34 \times 50k \times 32k$~55M bp 长度的序列。
数据组织格式可能是如下格式(每条数据):
{'region info': 'sample i, chr j, start k, end l', 'DNA sequence': 'ATAGCTA...TCC', #length 32k 'epi signal 1': [0,0.1,1.1,0,0,0,...,2.5,0,0], #length 32k # other epi signals 'epi signal n': [0,0.1,0.1,0,1,0,0,...,2.5,0,0]} -
分词:
对DNA序列, k-mer 分词, k=6, 特殊token [CLS], [MASK]
对epi 信号,离散分词,每个epi信号有36个token外加特殊token [MASK]
-
模型pipeline(以一条数据为例):
raw_data = {'region info': 'sample i, chr j, start k, end l', 'DNA sequence': 'ATAGCTA...TCC', #length 32k 'epi signal 1': [0,0.1,1.1,0,0,0,...,2.5,0,0], #length 32k # other epi signals 'epi signal n': [0,0.1,0.1,0,1,0,0,...,2.5,0,0]} ''' step 0: sample data ''' input = {'DNA sequence': 'ATAGCTA...TCC', #length L+k-1 (sample from raw_data['DNA sequence'][idx:idx+L], L \in {128,1024,2048,...,16384} 'epi signal 1': [0,0.1,1.1,0,0,0,...,2.5,0,0], #length L+k-1 # other epi signals 'epi signal n': [0,0.1,0.1,0,1,0,0,...,2.5,0,0] } # 上面使用 l+k-1的长度原因是因为k-mer 分词,分词时逐渐向右shift 1bp,最终分词的序列长度为 l ''' step 1: tokenization ''' # 分别处理 DNA-seq和 7个epi signal,得到token_ids,同时对需要mask的token进行mask, # token_ids.shape = (8, L), 这里的8是DNA seq + 7 个epi signal ''' steps 2: embedding ''' # 对 8 个token_ids分别进行embedding, token_embedding.shape = (8, L, d), d是embedding的维度 # sum token_embedding, 得到 multiomic_embedding.shape = (8, d) # reshape into 128bp bins, embedding.shape = (L/128, 128, d) # cat [CLS] and plus position embedding 1, final_embedding.shape = (L/128, 129, d) ''' step 3: transformer-1 block ''' # 20 layer transformer layer, embedding.shape = (L/128, 129, d) # for masked token, compute the classification CE loss, get loss_1, # extract cls token, plus position embedding 2, cls_embedding.shape = (L/128, d) ''' step 4: transformer-2 block ''' # 20 layer transformer layer, cls_embedding.shape = (L/128, d) # for each cls token, predict the preserved epi signal, classification CE loss, get loss_2 # pretraining loss = loss_1 + loss_2在整个训练过程中,对DNA seq 进行mask,需要预测mask掉的DNA seq,对7个epi signal, 将其随机分为两组,selected 和 preserved. 对selected 中的epi signal 进行mask,之后利用transformer-1 block中的输出预测masked token。 对preserved中的epi signal,使用transformer-2 block中的 L/128 个输出token进行预测,预测preserved中的每个bin的均值。
作者解释说这里两个transformer block的设计是为了抓住不同粒度的信息。transformer-1中是每个token(6bp)的粒度, transformer-2中每个cls token是128 bp的粒度。
从算法的角度来看,有点像block attention。将序列分成几块Block后,每个Block内部做self-attention, 之后block之间再做 self-attention.
通过Elementary 的训练,模型应该学到DNA序列表征和个体层面的表观信号序列表征。在这个基础上进行Regulatory的训练,在单细胞数据上,让模型学习到细胞层面上的表观特征。
CREformer-Regulatory
-
预训练数据:
使用10X multiome数据,来自10X官方的数据集。对每个细胞的每个基因,收集其对应的atac peak集合做为一条样本,共有165M条数据,可能的一条数据格式如下:
{'data info': 'cell i, gene j', 'gene expression': 1.1, 'peak set':{'peak 1' ['ATCG...TG', [0,0,1.1,0,...,0,1], '-123'], 'peak 2' ['ATGC...TG', [1,1.4,1.1,0,...,0,1], '+123'], # other peaks } } # 其中peak set 是全部 该 gene 的可能的peak 集合,对每条peak, # 记录序列信息,epi signal, 以及距离TSS的距离(+-表示上下游) # 每条peak,限制长度为1024bp -
分词:
同上
-
模型pipeline(以一条数据为例):
raw_data = {'data info': 'cell i, gene j', 'gene expression': 1.1, 'peak set':{'peak 1' ['ATCG...TG', [0,0,1.1,0,...,0,1], '-123'], 'peak 2' ['ATGC...TG', [1,1.4,1.1,0,...,0,1], '+123'], #other peaks } } ''' step 0: tokenization ''' # 分别处理每条peak的序列信息和 epi signal 信息,得到 (peak,2)条token id ''' step 1: CREformer-Elementary(frozen) ''' # 将上面的数据通过 CREformer-Elementary, 得到embedding.shape = (peak, 8, d) # 这里的 8 = 1024/128,因为每个peak的序列长度为1024,而Elementary中有划分128bp的bins ''' step 2: Attention pooling ''' # 这里需要汇聚全部peak的信息进行预测,使用attention pooling的做法 # (embedding + TSS-distance embedding).shape = (peak, d) ''' step 3: transformer block ''' # 20 layer transformer layer, embedding.shape = (peak, d) ''' step 4: zero padding ''' # 填充每条样本的embedding.shape 为 (peak_m, d) # peak_m是定值 ''' step 5: prediction ''' # 根据 peak_m*d 的embedding,预测基因表达值,MSE loss
以上为CREformer 模型的主要内容,模型总参数量为3b,从论文的附图中能看到数据量、参数量的增加对性能的提升。
结果1:zero-shot 推断 master TF 和 GRN
首先作者定义如下指标,对每个peak j, 定义Attention_Score(j)
\[\text{Attention Score}(j) = \sum_{i=1}^{L} \text{Softmax}\left(\frac{q_i \cdot k_j}{\sqrt{d_k}}\right)\]这里L是潜在RE的数目,猜测这里的潜在RE应该是该peak上下游窗口内的RE。Attention Score只使用CREformer-regulatory中的transformer计算。这里计算时仅需要序列信息。
由此,对每个peak,可以获得一个attention score,论文中发现这个attention score和 epi-signal高度相关。例如下图中b, attention score和三个master TF的 chip-seq signal高度相关。
同时作者对每个gene,定义了一个新的特征attention feature
\[\scriptstyle \text{Attention Feature}(G_iC_j) = \scriptstyle \text{Cosine Similarity}\left[\text{Attention Score}(G_iC_j), \text{Attention Score}(G_iC_{\text{max}})\right] \cdot \sum_{R_{tss}} \text{scATAC}_{G_iC_j}\]可以理解为这是一种新的gene activity的计算策略,在下图中的c中以及后面的dotplot中可以看到,这个新的特征能够很好的区分各个细胞类型的master TF(将 peak count转成 attention feature, 之后用logistic regression删选top feature)
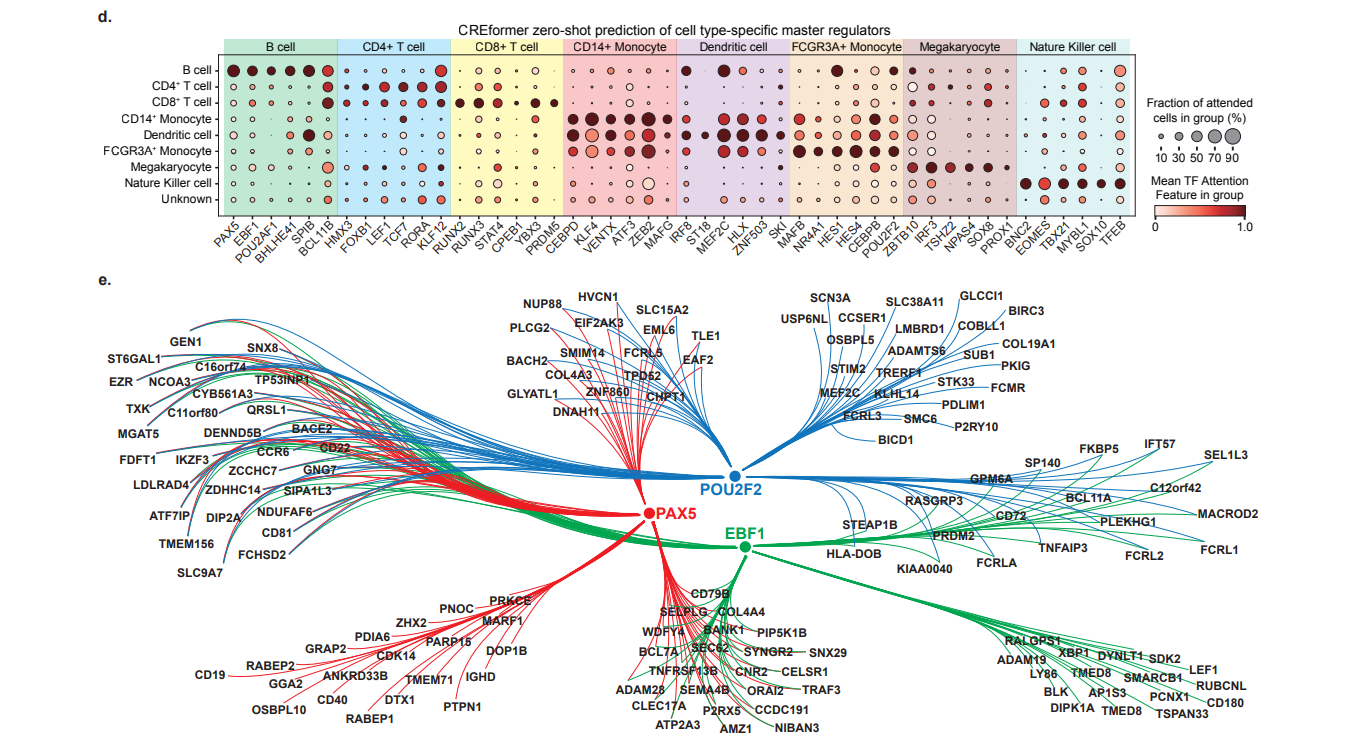
至于GRN的构建,对每个基因和每个TF,计算该基因的TSS上下游窗口中的,高attention score的peaks中的motif富集分数,设置阈值,建立TF->TG连边。
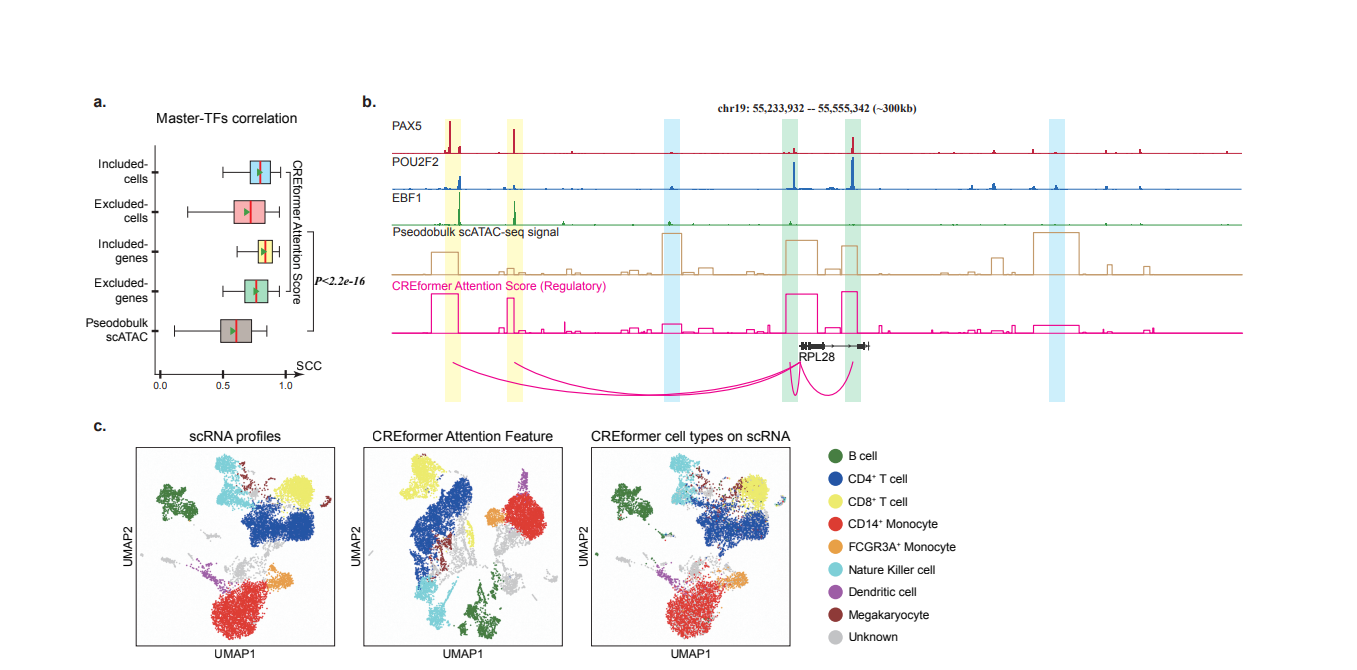
Remark 1:
基本逻辑:
- attention score 和 master TF的epi signal 高度相关
- attention score 高的peak 大概率是TF可以结合的调控元件
- attention feature 可以很好的区分各个细胞类型的master TF
- 对每个细胞类型构建GRN, 首先通过attention feature筛选出master TF。之后对每个TG,根据attention score筛选可能结合的peak。最后,对每个TF-TG pair, 计算每个TG窗口内的motif富集分数,阈值筛选,建立TF->TG连边。
看起来挺自洽的。。。做成TF-RE-TG三元组似乎也不难。但是评测未免有些草率,仅仅check了和SCENIC+ 推出的网络的一致性。总之还是缺少很好的GRN评测。但是GRN图很好看。
结果2:zero-shot 预测表观扰动
- 使用attention score 能筛选 enhancer-gene pair. 对每个基因,通过TSS窗口定义上下游潜在enhancer。之后对每个enhancer计算attention score, attention score的rank 中,实验验证的True enhancer-pair 会在前面。
- TF 敲除扰动预测:对每个TF,motif scan获取结合peak, 将结合peak的 atac signal信号赋0,之后CREformer-regulatory模型进行预测,获得扰动后的基因表达
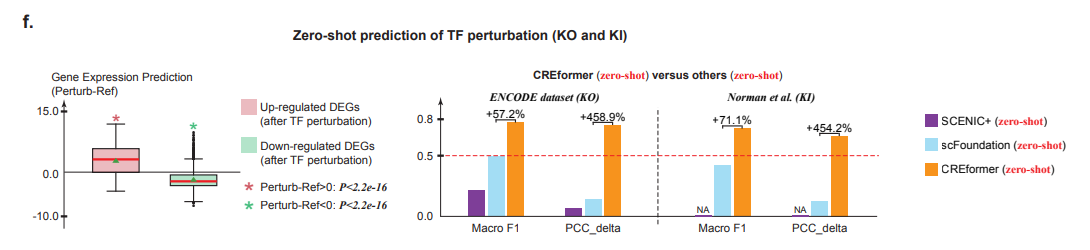
Remark 2:
对这些表观扰动的知识不是特别熟悉,结果看起来不错。但是TF扰动结果没有对比GET的结果,是GET做不了吗?看起来扰动结果只用了bulk 的ATAC-seq,而对比的两个模型是single cell的方法。
zero-shot 预测细胞状态转移
图a中展示了,对于发育到P0组织后上调的DEG,敲入这些转录因子后回下调这些DEG表达,可能恢复未发育状态。对下调的DEG回看到表达上调。
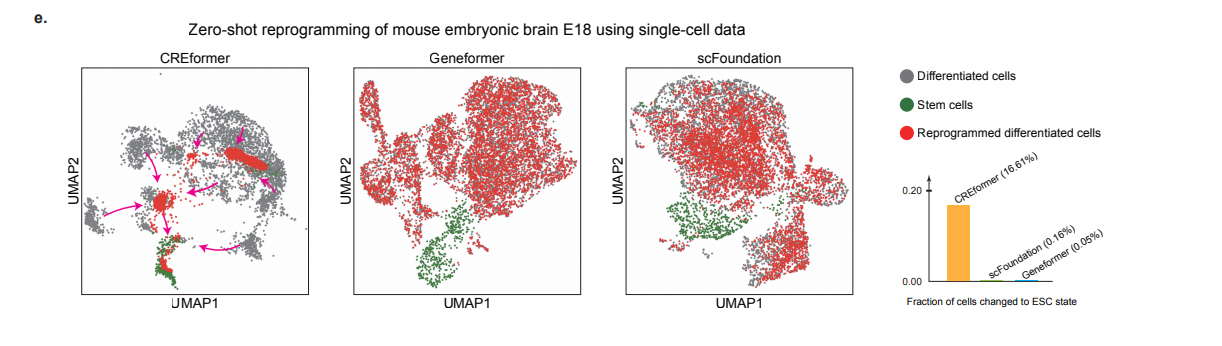
图b中展示了不同模型预测敲入TF后细胞状态变化,看起来CREformer预测的一致性最高。图画的有点费解,感觉没必要画这么复杂。
图e中展示了CREformer预测单细胞状态变化的工作。我们需要关注如下信息,绿色的是干细胞,灰色的是分化细胞,红色的是不同模型预测的TF扰动后的分化细胞. 在geneformer 和 scfoundation中,灰色红色完全重合说明预测的扰动后细胞和分化细胞基本一致,而CREformer能够看到灰色红色的差异,并且红色有部分和绿色的干细胞重合,说明CREformer预测的扰动后的细胞是有向干细胞分化的趋势的。
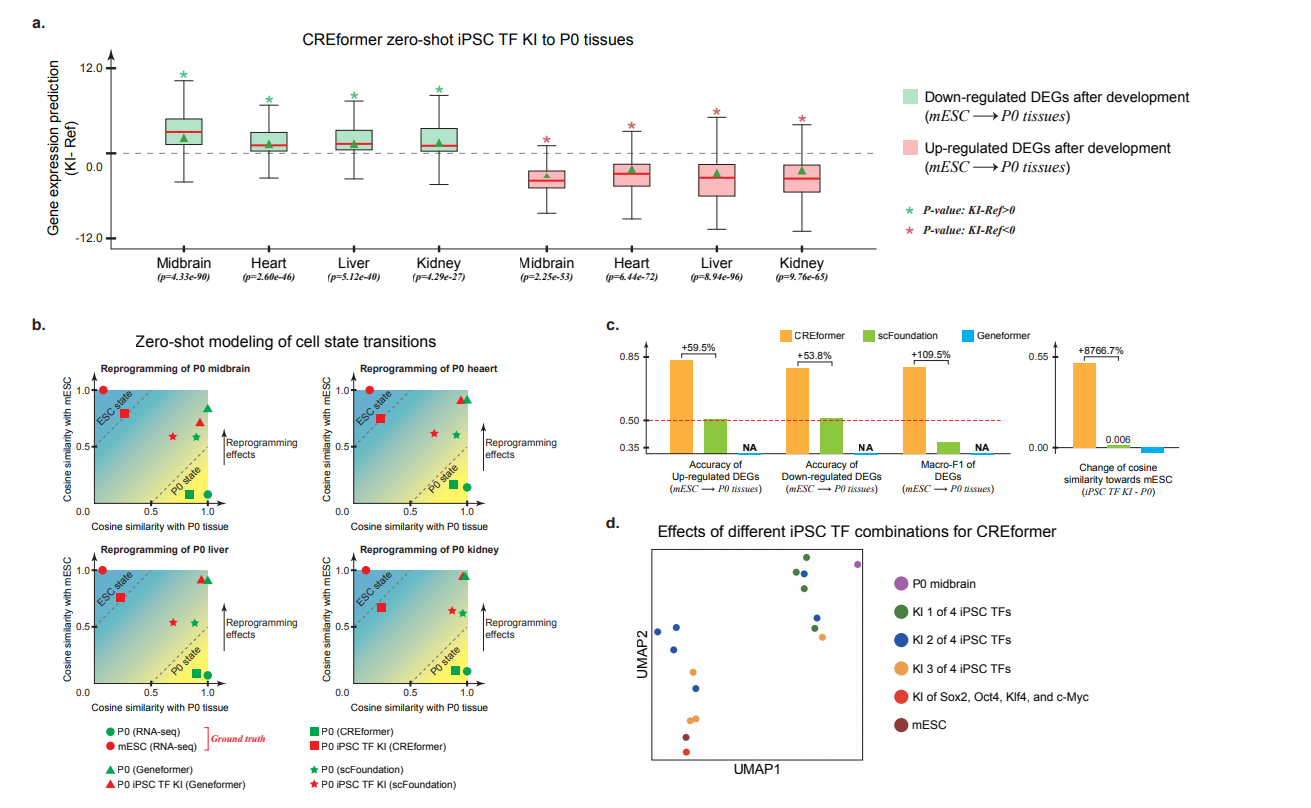
Remark 3:
细胞状态转移的实验设计的很巧妙,UMAP图做的也不错。在缺少额外证据的情况下,我愿意相信CREformer模型是有效的
zero-shot 寻找治疗靶点
有湿实验验证的结果,看起来很不错。首先基于Attention feature, 筛选正常细胞和肿瘤细胞的master TF, 并找到了乳腺癌的一个全新master TF. CREformer预测敲除该TF后可以促进肿瘤细胞向正常细胞转化。湿实验中也看到了敲除TF后,癌细胞增殖放缓。
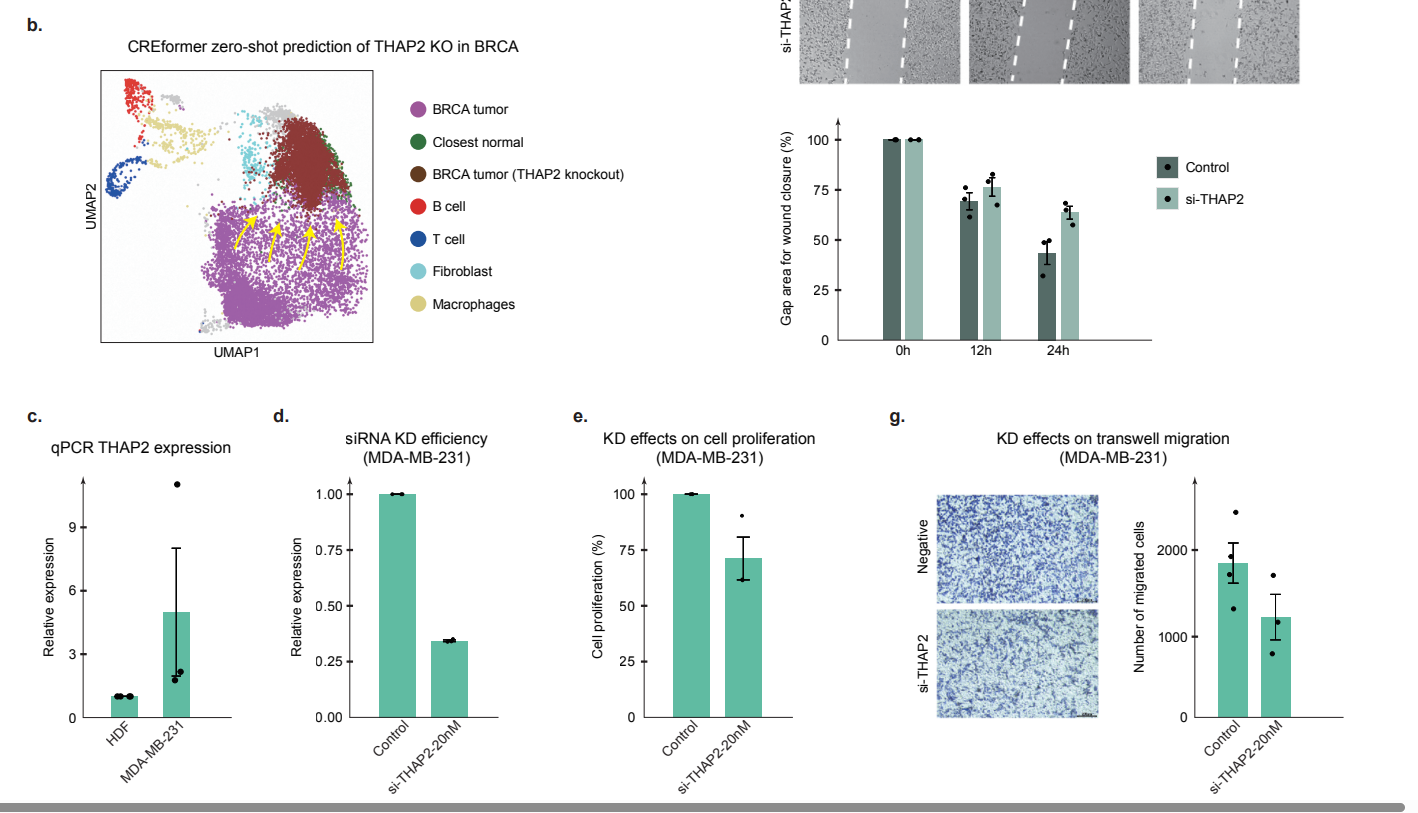
Remark:
- 实现了组学数据+序列数据的建模
- 几个比较有吸引力的结果,attention score和表观信号的高度相关性,这保证了可以利用模型的预测去推断一些表观的知识,例如TF结合peak。Reprogramming的结果,单细胞的UMAP展示没见过,新意上打满分。一个看起来不错的湿实验验证。
- 不清楚seq-embedding的收益点在哪里,似乎没有单独针对序列的任务
- 给我留了单细胞组学+基因组序列的建模空间哈哈哈~~